扩增子测序数据分析很难?一行命令实现从原始数据到分析报告的全自动化流程!
Dix-seq:一款基于模块化设计的扩增子测序数据全自动分析软件
Dix-seq: A one-step amplicon sequencing data analysis software designed with modular design.
Dix-seq:一键完成扩增子测序数据分析软件
【全文亮点主旨总结】
Dix-seq offers a one-step pipeline to complete the entire amplicon sequencing data analysis.
Dix-seq supports advanced customizations through its modular design.
Dix-seq generates retrospective scripts, makes reproducing results or debugging errors easy.
【初衷】
扩增子测序作为开展微生物组学研究不可或缺的工具之一,具有成本低廉,受宿主污染影响较小,交付周期较短的特点。然而,目前开源免费的扩增子测序数据分析软件使用起来仍较为繁琐,给一线科研人员带来了不小的挑战。因此,开发一款完全开源、易于部署、使用简单的扩增子测序数据分析软件有助于降低扩增子测序技术的应用门槛,并进一步推动扩增子测序乃至微生物组学方法在更多研究领域的应用。
【正文内容】
导读
高通量DNA测序的成熟极大地推动了微生物组学的发展。然而,扩增子测序作为开展微生物组学研究的首选工具之一,仍存在着数据分析较为繁琐复杂的挑战。因此,我们开发了Dix-seq:一款能全自动完成扩增子数据分析并生成报告的软件,以降低该技术的应用门槛,并推动微生物组学方法在更多领域的应用。
Cover Page
正文
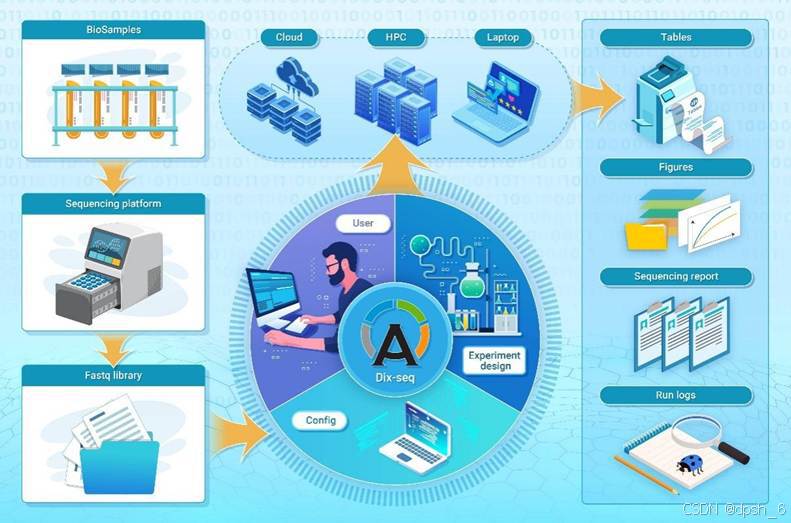
过去十年间,测序技术的飞速发展推动了扩增子宏基因组学的广泛应用。然而,当前的扩增子数据分析软件/流程往往需要在多个步骤中进行人工调整,用户需要对软件中的参数有较为全面的了解,这给初次接触扩增子测序数据分析的一线科研人员带来了不小的挑战。因此,我们开发了Dix-seq,一个用于快速、自动化、可扩展并完全容器化的扩增子数据分析工具。Dix-seq可以一步完成从原始测序序列处理到多种统计分析和可视化等工作,并生成基于网页的分析报告和回顾性日志文件(图1)。Dix-seq使用单一参数卡机制,可以明显简化其命令行界面,研究人员只需填写好实验设计表格,并将项目信息填写进参数卡配置文件中即可开始分析,使非专业用户更容易上手;另外,通过参数卡记录分析参数,也可以提高研究的可重复性(图1)。最后,Dix-seq的模块化设计使其能够快速采用新的方法和数据库,以融入其软件框架。目前,已有超过21种算法、软件和第三方程序被整合到Dix-seq的八个模块中,涵盖了从下机数据拆分到下游常规统计分析绘图等常见分析步骤(图1);后续更新将会有更多数据分析方法和数据库加入到Dix-seq的自动化分析流程。对于有数据分析经验的科研人员,Dix-seq提供了自定义分析功能,用户不仅可以对其数据分析模块流程进行组合调整和单独调用,还能快速封装自己的分析模块,以更好地为用户提供个性化分析服务。
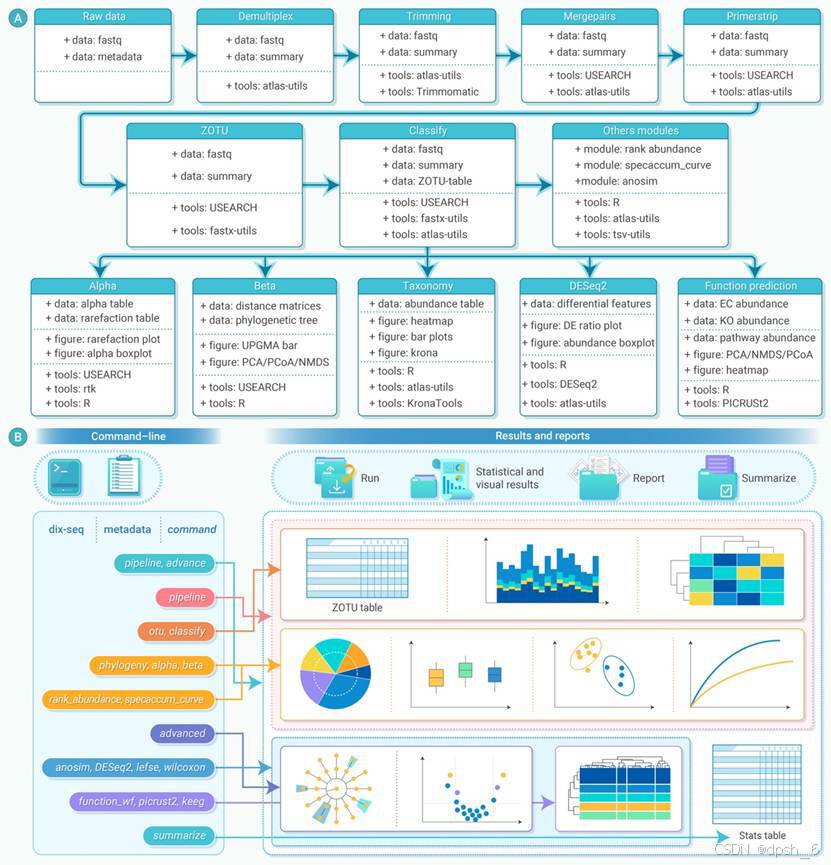
图1 Dix-seq:快速扩增子数据分析的一体化流程。(A)Dix-seq流程中使用的步骤和软件。(B)通过Dix-seq中的“pipeline, advanced”一步式工作流程完成整个分析的概览示意图,以及用于自定义分析的Dix-seq模块化设计等。
在真实案例研究的数据集上进行的基准测试表明,Dix-seq可以生成包含统计信息的出版级图形和对应的数据信息表(图2-3)。
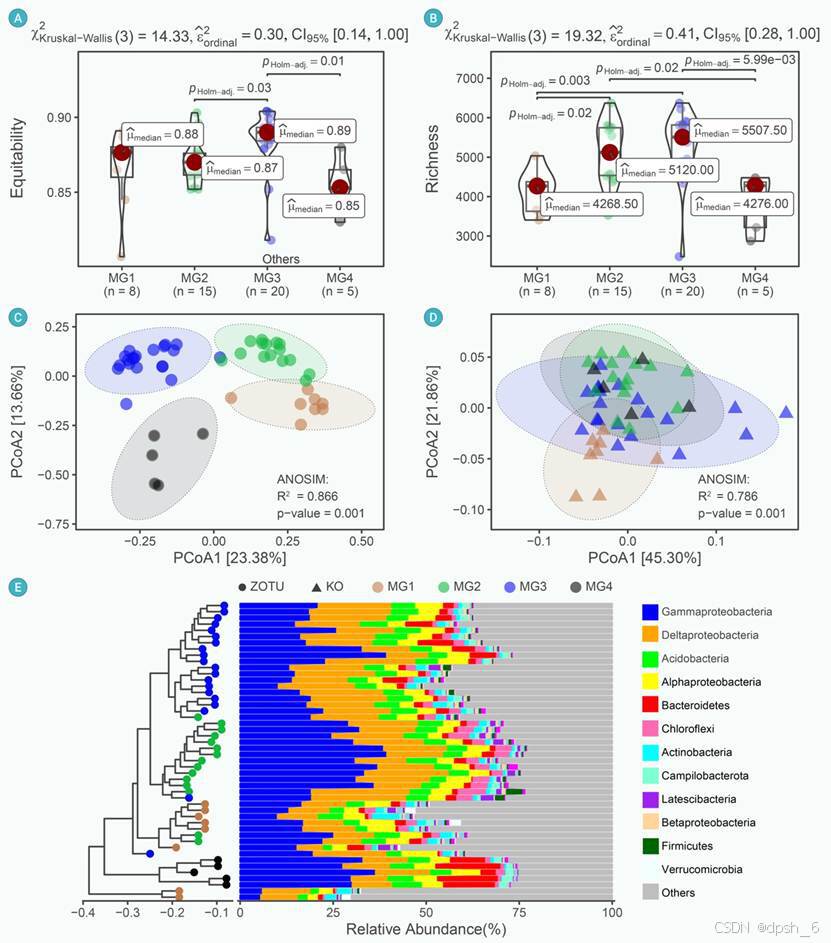
图2 通过使用Dix-seq中的一步式工作流程进行研究案例I(Wei et al., 环境微生物组)的分析和可视化结果,展示了长江河口区域微生物类群(ZOTUs)和功能(KOs)的差异。(A和B)小提琴箱线图展示了四个区域中微生物α多样性指数【包括均度/均匀度(A)和丰富度(B)】的差异和概率分布。使用Kruskal-Wallis检验计算统计差异,并使用“holm”方法计算校正后的P值。框内的红点分别代表中位数。框顶部和底部的边缘分别代表1.5倍的四分位数间距(IQR)。(C和D)基于Bray-Curtis相异性的主坐标分析(PCoA)可视化了48个样本在四个河口区域的ZOTU(C)和KO(D)组成的变异。通过相似性分析(ANOSIM),证实了四个地理区域之间的微生物群落结构和功能组成变异存在显著差异。(E)基于加权UniFrac距离的UPGMA系统发育树,结合了门/变形菌纲水平的物种分布。不同沿海区域的样本数量如下:MG1(n=8),MG2(n=15),MG3(n=20),MG4(n=5)。
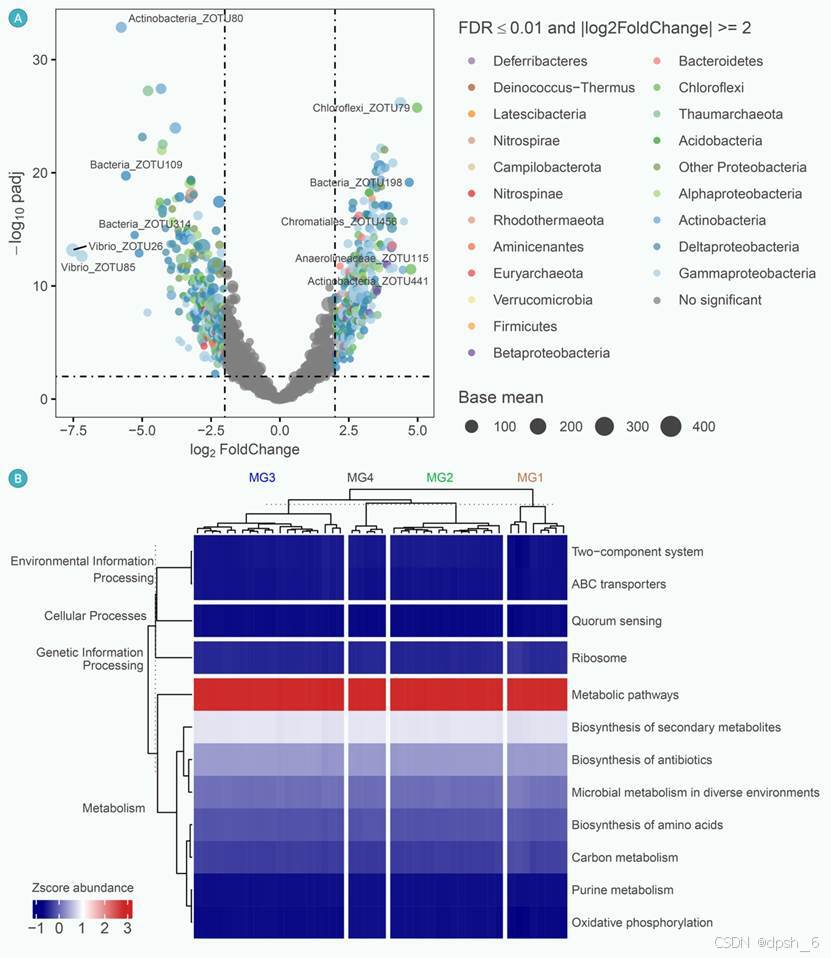
图3 使用Dix-seq分步式工作流中的组合子命令 “DESeq2,picrust2,keeg” 的可视化结果示例。(A)比较MG2和MG3组之间的差异ZOTUs。圆的直径与给定细菌ZOTU的平均丰度成正比。选择|Log2(FoldChange)| > 2和错误发现率(FDR)≤ 0.01的ZOTUs作为差异ZOTUs,并根据其分类学用不同颜色绘制。(B)热图展示了四个河口区域微生物群落功能预测结果中,前12个最高丰度的功能通路分布情况。通过调用第三方“PICRUSt2”软件对KEGG 2级类别的丰度进行归一化,并在Dix-seq中使用Z分数进行缩放。
此外,我们利用测试数据集2(Wang et al.),将Dix-seq生成的结果与当前主流扩增子数据处理流程进行分析比较。在群落多样性与物种注释(图4)、群落结构(图5)和群落变异解释度(图6)等相关结果中,Dix-seq在所有测试项目中均表现靠前。
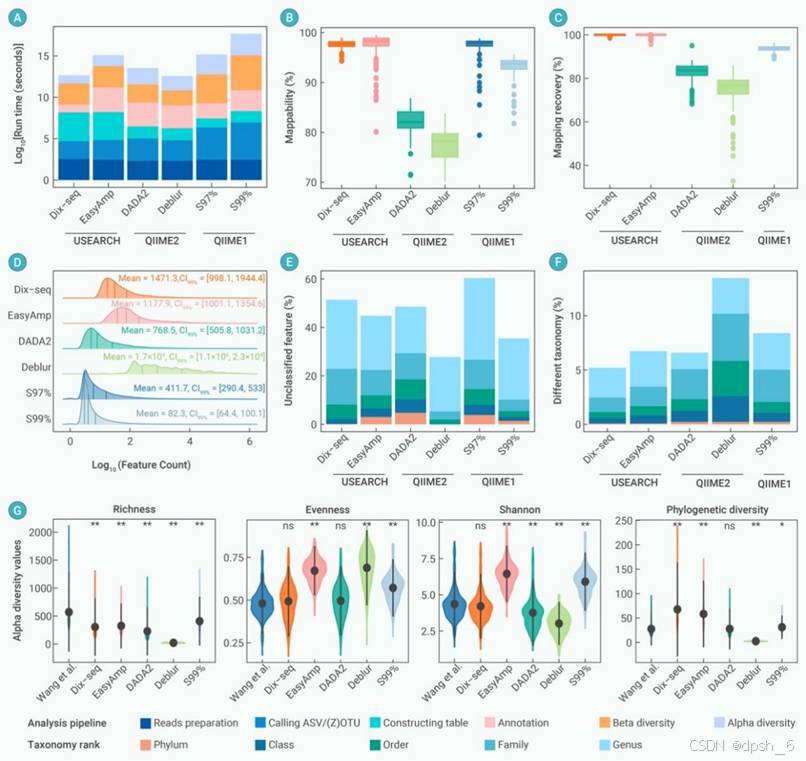
图4 使用不同的分析流程和策略对研究案例II(Wang et al.)数据集的运行时间、序列利用率、物种丰度分布、物种注释和α多样性指数进行比较。(A)比较微生物组分析的推荐方案与六种方法的所用时间。这些方案进一步分为六个步骤(analysis pipelines):1. reads准备,2. 生成ASVs/ZOTUs,3. 特征表构建,4. 物种注释,5. β多样性,6. α多样性。(B和C)使用多种数据分析方法和策略用于比较原始序列的利用率。可映射性(Mappability)是通过每个样本未过滤特征表中保留的原始读数的百分比进行计算(B)。将研究案例II中公开发表的代表性序列,与其他分析策略生成的序列进行比对,以评估序列映射恢复率(Mapping recovery)的差异(C)。(D)脊线图揭示了6种分析策略产生的特征表中ZOTUs/ASVs丰度的概率分布。箱图中的直线分别代表中位数(中心线)、上四分位数和下四分位数。(E和F)多种分析策略的物种注释结果比较。比较6种分析策略结果中,未分类单元ZOTUs/ASVs的平均丰度占比(E)。在五类物种分类等级(门、纲、目、科和属)上,研究案例II的已发表结果与其他数据分析策略的分类结果的差异比较(F)。(G)小提琴图展示了研究案例II中,已发表的微生物α多样性的结果和其他5种分析策略之间的差异。星号表示Wang et al.已发表结果与其他五种方案之间的统计显著性(“*”,P < 0.05,“**”,P < 0.01,Kruskal-Wallis检验)。(C,F,G)用于比较案例II已发表结果与其他五种方案在代表性序列、分类学和α多样性方面的一致性。易扩增子(EasyAmplicon)简写为EasyAmp。
-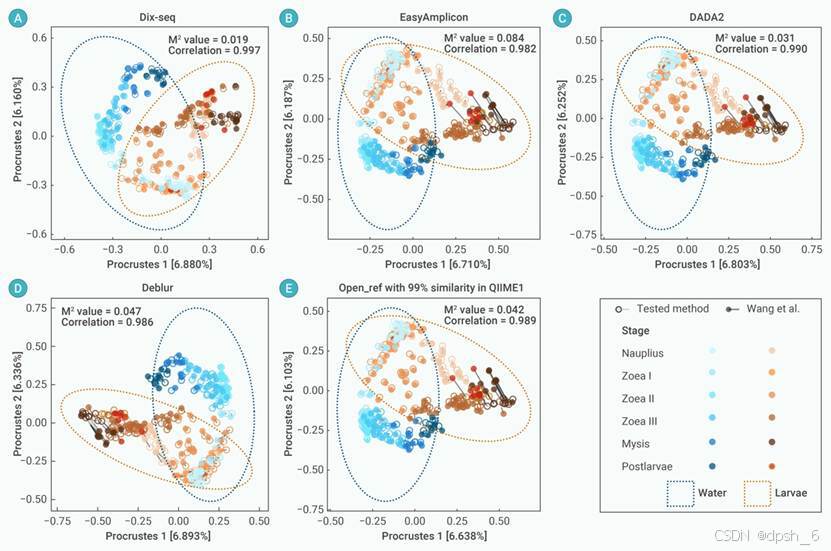
图5 使用普氏分析(Procrustes analysis)比较分析5种数据分析策略的群落结构结果与案例II(Wang et al.)已报道结果的相关性差异。普氏分析的数据基于ZOTUs/ASVs表的Bray-Curtis相异性矩阵进行。样本(点)根据对虾幼体的发育阶段和微生物群落的栖息地类别分别进行配色。相关性和M2分别代表两个矩阵之间的Procrustes相关性和平方和。M2值越接近0,表示案例研究II(Wang et al.)的群落结构结果与被测试方法间的一致性越高。通过999次排列估计Procrustes统计的显著性,所有P值<0.01。
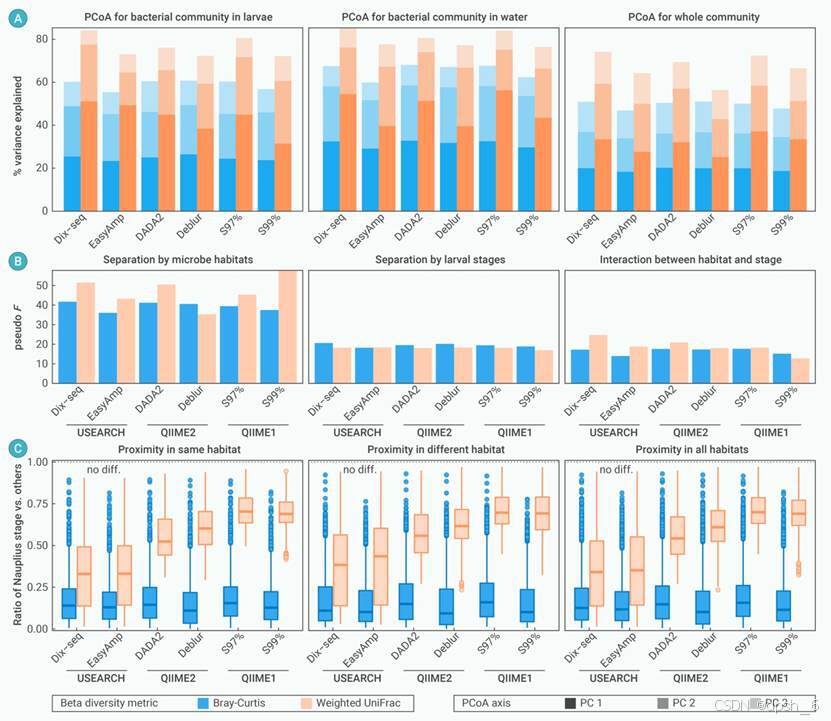
图6 使用6种数据分析策略对研究案例II(Wang et al.)的数据集进行β多样性分析结果的比较。(A)在不同栖息地类别(左:仅在幼体肠道,中间:仅在养殖水体,右:总微生物群落),微生物群落结构PCoA降维分析结果前三个轴的解释度比例。(B)使用置换多元方差分析(PERMANOVA)检验栖息地(左)、宿主发育时期(中)及双变量的相互作用(右)对微生物群落结构是否有显著影响,并统计pseudo F值。(C)溞状幼体(对虾发育早期)阶段微生物组成与其他幼体发育阶段的差异比较。该差异比值的详细计算方法请参考原文。该比率越接近1,表示溞状幼体阶段的微生物群落结构与其他阶段之间的差异越低。
在模拟测序深度降低时,Dix-seq在检测群落变异方面仍然保持高度有效性,尤其是在系统发育多样性和皮尔逊相关性等结果中,即使在低测序深度时,Dix-seq的分析结果仍保持稳健(图7)。综上所述,Dix-seq是一个既方便又可灵活定制的扩增子数据分析工具,是初级和高级用户的理想选择。
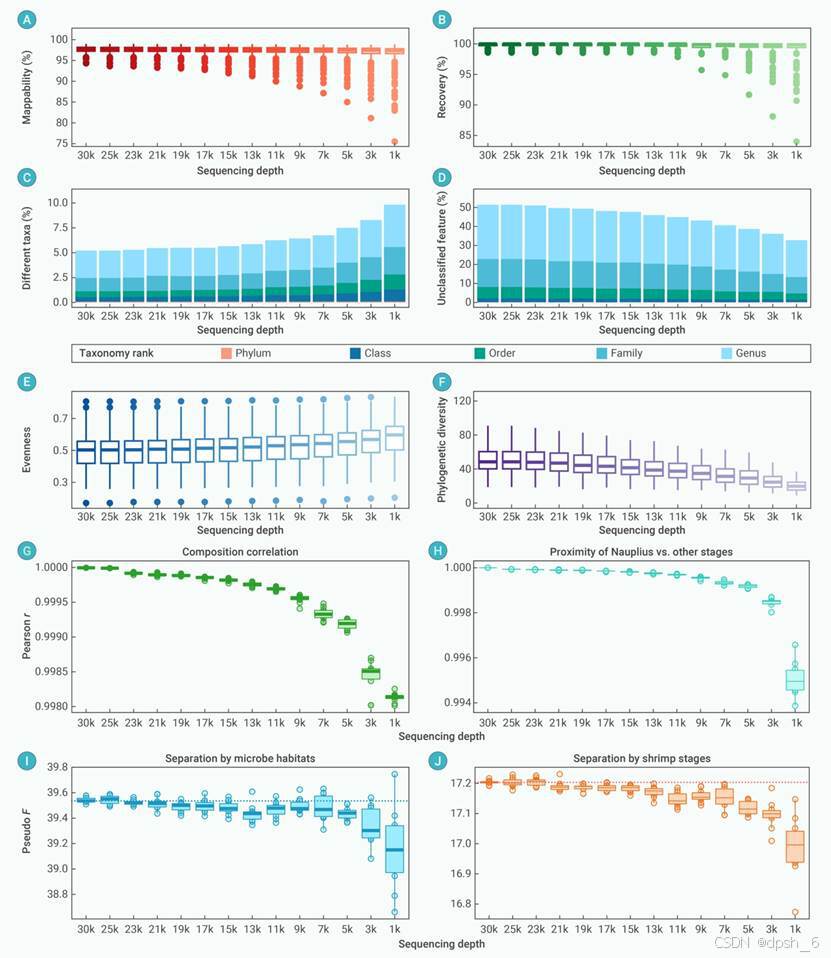
图7 测序深度对Dix-seq软件结果的影响。(A和B)Dix-seq的可映射性、映射回复率与样本测序深度之间的关系。(C和D)使用Dix-seq软件时,物种注释差异分类群、未分类类群与样本测序深度之间的关系。(E和F)Dix-seq软件分析结果中的均匀度(E)和系统发育多样性(F)等α多样性与样本测序深度之间的关系。(G)原始ZOTU表与基于不同测序深度值的随机抽样表之间的皮尔逊相关系数(r)。(H)溞状幼体阶段的ZOTU表距离矩阵的平均比率与处于其他发育阶段平均比率的皮尔逊相关系数(r)随不同样本测序深度的变化情况。(I和J)使用置换多元方差分析(PERMANOVA)检验不同样本测序深度下,栖息地(I)和对虾幼体发育阶段(J)对微生物群落结构是否有显著影响,并统计pseudo F值。
Dix-seq的安装与使用说明
Dix-seq(下载地址点我)支持通过Apptainer(安装方式如下:Ubuntu, RHEL)容器化运行。在开始分析之前我们还需要准备以下内容:
1. 参考数据库(下载地址点我)
2. 实验分组表格
3. 配置文件
首先重命名fastq文件,样本命名需要符合分析规范(样品命名请以字母开头,限数字、字母和“-”,名称长度不超过 10 个字符,对于 Illumina双端测序平台,每个样本包括R1和 R2 端两个测序文件), 样本名称模式:样本名称_1.fastq 或者 样本名称_1.fq,可以使用 gz 压缩文件格式,将所有重命名好的fastq文件存放在raw_data文件夹下。
接下来准备实验分组表格(mapping_file.txt),共两列,制表符分隔,第一列为样本名称,第二列是分组信息
#SampleID Description
A-1 A
A-2 A
A-3 A
B-1 B
B-2 B
B-3 B
最后修改配置文件(metadata.txt,模板文件点我下载),一般只需要改动下面这一小部分
project_id <输出结果文件夹的名称>
cpus <多进程并发数量>
parallel <并发任务数量>
threads <多线程数量>
这里可以根据硬件性能酌情修改
mergepairs_param -fastq_minmergelen 350 -fastq_maxmergelen 530 -fastq_pctid 90 -fastq_minovlen 16 -fastq_trunctail 2 -fastq_minlen 64
search_pcr_param -fwdprimer <5’ 端引物序列> -revprimer <3’ 端引物序列> -minamp 350 -maxamp 500
这里如果是V3-V4多样性测序只需要填写引物序列即可,如果测的是其他可变区还需要根据目标扩增片段长度修改-fastq_minmergelen,-fastq_maxmergelen,-minamp,-maxamp这几个参数。
db <填写参考数据库的所在文件夹地址绝对路径(如:/biostack/dix-seq/db)>
reference <填写参考数据库的文件名(不带拓展名:如silva-138.2.ssu_nr99.udb这里填silva-138.2.ssu_nr99)>
这里可以设置物种注释所使用数据库名和地址,数据库可以在figshare进行下载, https://doi.org/10.6084/m9.figshare.27978744.v2;或使用以下链接(可全部复制到浏览第地址栏)下载:
https://efile111.hpccube.com:65014/efile/s/w/Ymlvc3RhY2s=_cb8e382fb31e6b53&(密码:kXTz)请在浏览器打开链接,输入提取码下载文件?pwd=kXTz
准备工作完成后就可以使用以下命令运行Dix-seq
apptainer exec --bind <数据库文件夹路径,如:/biostack/dix-seq/db> <Dix-seq镜像文件存放位置> dix-seq metadata.txt pipeline,function_wf,advanced,summarize,report展望
大数据时代下微生物组学的发展迎来了新的机遇与挑战,因此我们开发了Dix-seq以便于用户处理扩增子测序数据。测试结果表明Dix-seq的综合性能基本上略强或持平当前主流的扩增子测序数据分析软件,而且其部署方便,使用简单,可拓展性强的优点不仅能够帮助经验不足的用户快速分析,还能帮助经验丰富的用户定制分析流程。总之,我们希望Dix-seq能够进一步推动扩增子测序技术的应用。
原文链接:
Dix-seq: An integrated pipeline for fast amplicon data analysis
【作者简介】
董鹏生,河南农业大学动物科技学院讲师,2022年3月博士毕业于宁波大学获水产养殖专业。主要从事对虾肠道微生物与宿主互作机制等研究。开发了一系列用于处理微生物组大数据的生物信息学算法和数据库系统,为以肠道微生物组为核心的对虾绿色养殖和健康精准管控提供了方法和理论依据。主持国家自然科学基金青年基金、河南省科技攻关项目、河南省“三区人才”项目等,参与国家863项目、国家重点研发计划项目、国家自然科学基金面上项目等省部级以上项目5项。在Microbiome、npj Biofilms and Microbiome、Molecular Ecology、Aquaculture等国际主流SCI刊物上发表学术论文10余篇;授权软件著作权4项,其中1项转让到企业。担任npj Biofilms and Microbiome、Aquaculture、BMC Microbiology等期刊审稿人,荣获宁波市自然科学优秀论文奖(B等)1项,指导学生荣获第二届高校大学生水产类创新实践能力大赛一等奖、第八届全国大学生水族箱造景技能大赛三等奖等奖励6项。
魏勇军,郑州大学药学院副教授,博士生导师,河南省中兽药发酵制剂工程技术研究中心主任。获得第十五届河南青年科技奖等。主要研究方向:中草药活性分子的微生物转化和合成。主持国自然面上、国际(地区)合作与交流项目、青年基金、河南省优青、河南省高校科技创新人才等项目。已发表论文90余篇,其中,以第一/通讯作者在Trends in Biotechnology、iMeta等期刊发表论文60余篇。以第一主编在Elsevier出版合成生物学英文书籍2本,主编/参编中英文著作6本。担任BMC Microbiology、iMeta、The Innovation等30余本英文SCI期刊编委、青年编委、客座编委等。担任Nature Communication等60余本高水平杂志的审稿人。已获批24项国内外专利,在审专利16项。相关成果获得河南省科技进步奖二等奖、河南省生物工程学会2024学术年会论文特等奖、第二届全国大学生化妆品创新大赛优秀指导教师等。担任国际应用微生物学会干净水科学咨询委员会委员、河南省微生物学会理事等。指导国家级大学生创新创业训练项目等本科生科研项目9项;指导学生获得第二届全国大学生化妆品创新大赛一等奖、国家奖学金等20余项奖励。
李明,博士,教授,博士生导师,现任河南农业大学动物科技学院院长;中国畜牧兽医学会高级会员、养兔学分会副理事长;中国畜牧业协会兔业分会常务理事、专家团成员;科技部科技项目评审专家库专家;教育部本科教育教学评估专家库专家;河南省学位委员会农学类学科评议组成员;河南省畜牧兽医学会常务理事;河南省兔业分会理事长、专家团团长;河南省养猪学分会常务理事;河南省动物遗传资源委员会主任委员。河南省遗传学会动物与微生物遗传专业委员会委员。主要从畜禽遗传资源评估与利用,水产特种经济动物的养殖等方向的研究,主持完成包括国家重点研发计划课题、河南省现代农业技术体系、科技攻关、科技成果转化、科技扶贫等项目27项;发表SCI等学术论文125篇,其中第一/通讯作者65篇。主编和参编“十一五”、“十二五”规划教材等著作17部,获国家技术发明二等奖、国家教学成果二等奖、河南省科技进步一等奖等国家和省部级奖项6项。
张磊,2007年毕业于电子科技大学,先后在国家人类基因组南方研究中心、中国科学院上海植物生理生态研究所从事生物信息数据分析工作;2013年创建上海逻捷信息科技有限公司,参与生物信息工具和工作流的开发,在微生物组领域开发过近百个生物信息数据分析工作流,封装了超过6,000个生物信息相关容器,在Nature、ISME J、Cell Res、Water Research等多个国际著名期刊发表相关文章。围绕宏基因组学数据分析,根据不同需求开发过:meta-seq、reps-meta、mag-kit、vitg-kit、meta-inspect、bact-ann和bact-typing等工作流。

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐



 36
36 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)