EVBioX 第三期 | 单细胞数据清洗与处理全攻略
本期 EVBioX 聚焦单细胞测序原始数据的清洗与预处理,系统梳理了不同测序技术的原理、常见数据格式以及数据质控与标准化的关键环节,帮助大家打下高质量单细胞分析的坚实基础。
“单细胞测序的数据处理,如同精雕细琢的工艺过程,每一步精细打磨,方能成就完美成果。”
前两期中,我们聊过了单细胞分析的学习路线与方法技巧,但具体到真实的数据分析,我们首先面临的问题就是:如何正确处理和清洗庞大的原始数据?
本期,我们将系统而深入地讲解单细胞测序原始数据的特性、清洗处理的必要性、关键步骤和实践工具,帮助大家从源头就把控好数据分析的质量。
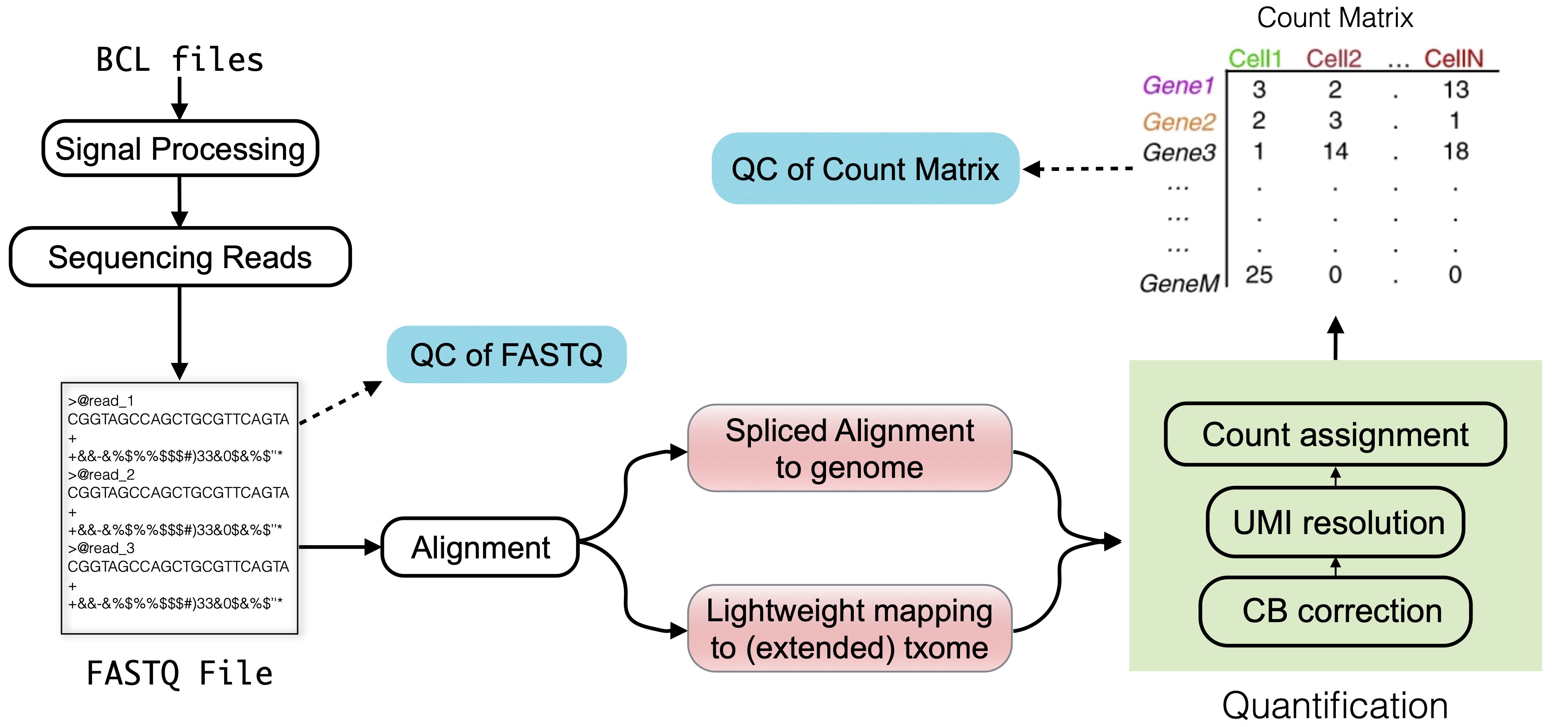
Fig. 3.1 An overview of the topics discussed in this chapter.
1. 单细胞测序种类与原始数据格式
1.1 常见测序类型与原理特点
目前常用的单细胞测序方法主要包括:
1.1.1 10X Genomics:
基于微流控技术的高通量单细胞捕获方法。细胞悬液通过微流控芯片产生微滴,每个微滴包含单个细胞以及带有细胞特异barcode和UMI标签的磁珠。通过反转录(RT)过程,细胞内mRNA与磁珠上的barcode和UMI结合,从而区分每个mRNA来自哪个细胞(barcode)以及去除PCR扩增过程产生的重复序列(UMI)。该技术具备高通量(一次捕获数千个细胞)、操作简单、分辨率适中的特点,适合大规模样本的异质性分析。
1.1.2 Smart-seq2/3:
不同于10X技术的微滴系统,Smart-seq系列方法基于单细胞手工捕获或流式细胞仪(FACS)分选。每个单细胞独立放入一个微孔板内进行逆转录反应,合成cDNA后再逐一文库构建并测序。因为每个细胞单独处理,所以获得的覆盖基因更广泛、测序深度更高,具有极高的分辨率,但由于成本高,适合小规模、精细化研究,如稀有细胞类型研究。
Fig. 3.2 An overview of the scRNA-seq technology.
不同技术的异同与应用场景
| 平台 | 代表方法 | 通量 | 灵敏度 | 是否全长 | 优势 | 适用场景 |
|---|---|---|---|---|---|---|
| 微滴法 | 10x, Drop-seq | 很高 | 中等 | 否 | 高通量,成本低,操作简便 | 大队列,罕见细胞探索 |
| 微井法 | Smart-seq2 | 低~中 | 很高 | 是 | 检测全长/低丰度/剪接变体 | 转录本结构/剪接/深度挖掘 |
| 杂合与新方法 | Seq-Well等 | 中等 | 中等 | 视具体平台 | 介于两者之间,适合特定样本 | 特殊需求 |
1.2 原始数据的格式说明
单细胞测序后产生的初始数据是FASTQ文件,记录测序得到的序列和相应的质量分数。
此外,每条序列都携带两个关键信息:
1.2.1 Barcode(细胞标签):
Barcode来自于逆转录过程中引入的人工合成的短寡核苷酸序列,用于标记单个细胞来源。不同barcode对应不同细胞,这使得每条序列可以明确区分来自哪个细胞。
1.2.2 UMI(Unique Molecular Identifier,唯一分子识别码):
同样是在逆转录步骤添加的一段随机的短序列,用以识别单个mRNA分子,去除PCR扩增带来的偏差。UMI的值代表原始mRNA分子的独特标记,能确保我们统计基因表达时避免过多计数PCR重复扩增片段。
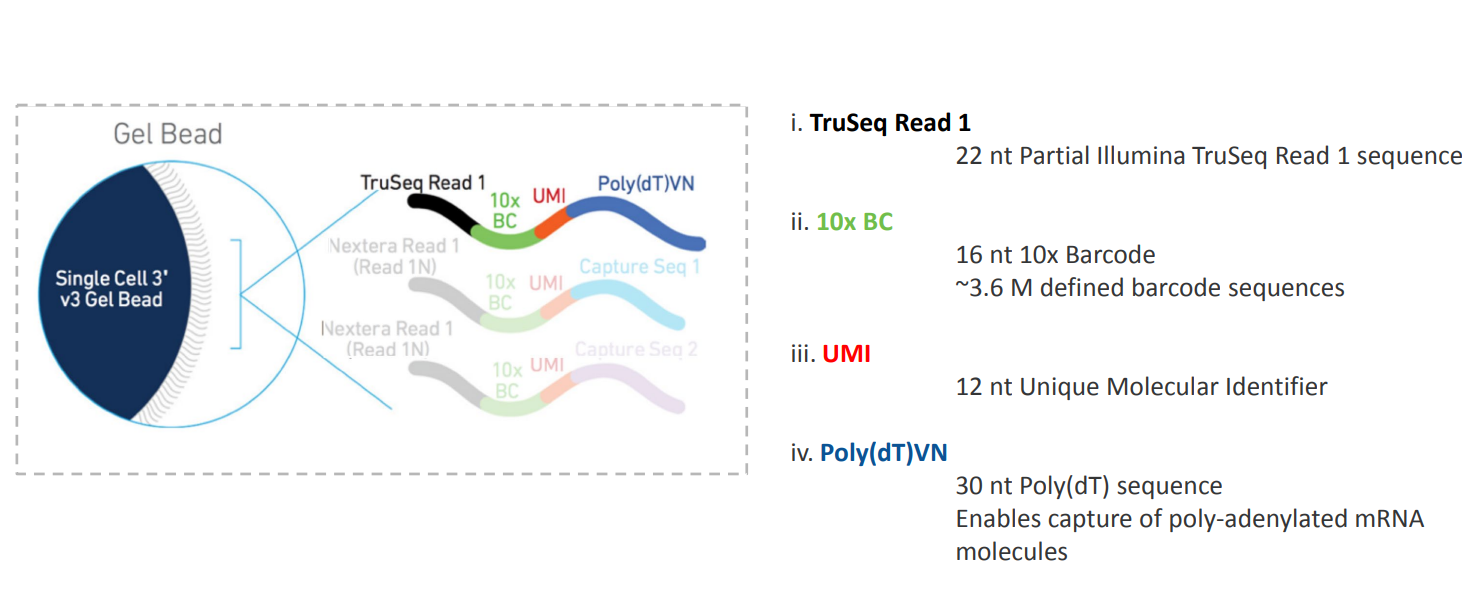
Fig. 3.3 Schematic diagram of cell barcode and UMI structure in single-cell RNA-seq libraries.
1.3 实际获取数据与初始格式
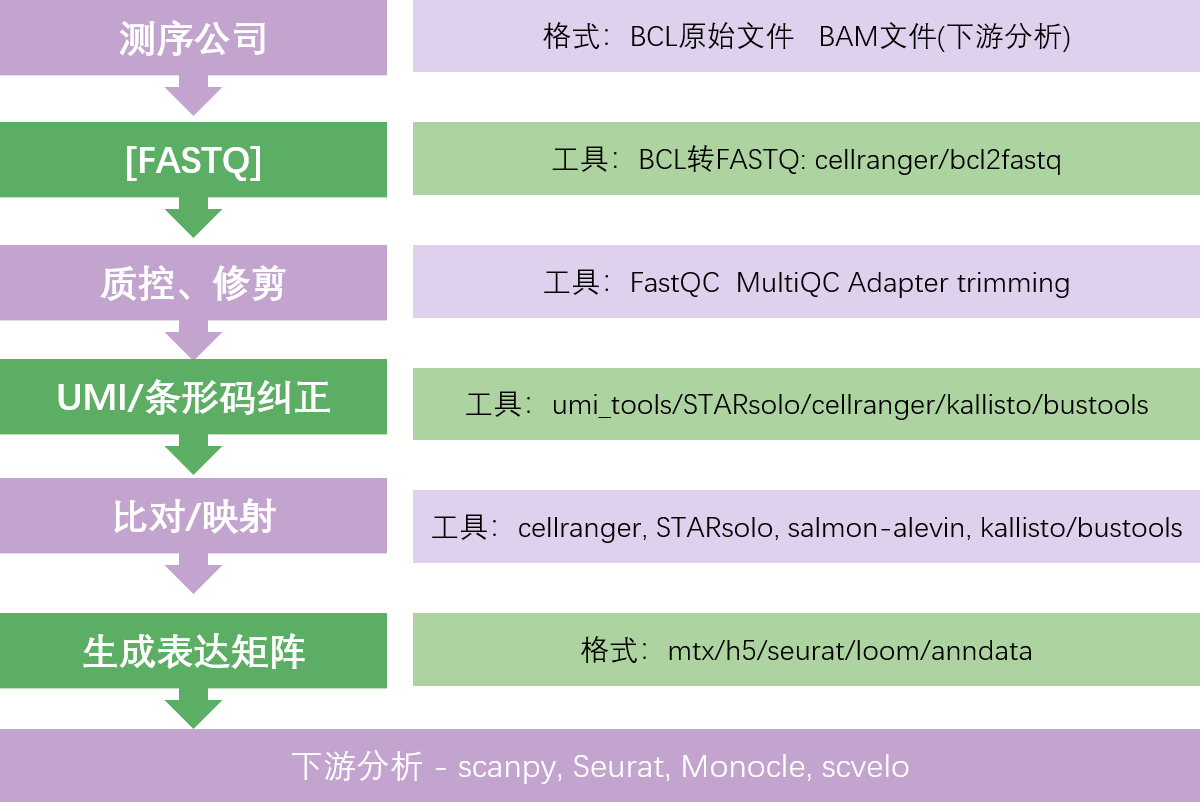
在完成单细胞测序实验后,测序公司(如10X Genomics, 华大、诺唯赞等)通常会直接为你提供原始测序文件,格式主要是FASTQ。部分公司还可能提供BAM、MTX等文件,或分析管道输出的中间文件夹(如Cell Ranger输出的“outs”目录,含feature-barcode-matrix等)。
-
FASTQ文件:存储原始reads及其测序质量,是所有分析的起点。
-
BAM文件:有时测序平台会同步交付比对后的BAM文件,它记录了每条reads与参考基因组的对齐信息。BAM既可直接用作部分分析(如RNA velocity/loom文件生成),也为QC等二次分析提供便利。
-
Feature-barcode matrix:部分平台(如10X)还直接提供稀疏表达矩阵,可直接用于Seurat/Scanpy分析。
FastQC报告解读:FASTQ文件拿到后建议先用FastQC检查reads质量。关注如“Per base sequence quality”“Adapter Content”等模块,判别是否需要修剪(trimming)。
BAM文件:比对后产生,常用于可视化、RNA速率分析(如velocyto生成loom文件)等二次处理。
2. 为什么需要数据清洗与预处理?
2.1 数据清洗的学术意义与目的
2.1.1 减少技术噪音和误差
测序数据中天然存在测序错误、PCR扩增偏差、低质量序列(如带Adapter序列),数据清洗可大幅减少这些技术误差,提高数据的可靠性和可复现性。
2.1.2 提高后续分析的准确性
干净的数据能更真实地反映细胞表达特征,避免误导后续的差异表达、聚类和降维分析。
2.2 数据清洗与处理的核心步骤
具体包括以下几个关键步骤:
1. 质量评估与控制(Quality Control,QC)
测序后,首先要对FASTQ文件进行质量检查,通常利用FastQC或MultiQC工具评估序列的整体质量(如碱基质量分布、测序饱和度、GC含量),明确数据整体质量和潜在问题。就像体检报告,告诉你哪些数据是健康的、哪些需要“治疗”。
2. Adapter去除和低质量序列修剪(Trimming)
测序片段常带有测序Adapter序列,这些序列必须用Trimmomatic或Cutadapt等工具去除。同时也去除序列两端低质量的碱基,避免影响后续比对和表达量统计。去掉序列中的“杂草”,确保后续分析数据的纯净。Cell Ranger 内部自动完成适配子去除和必要的质量过滤,无需用户单独处理。
3. Barcode和UMI的识别与修正
通过软件(如Cell Ranger或STARsolo)识别序列中的barcode与UMI,这一步需要对barcode和UMI进行错误修正(如单碱基错误容忍),确保正确统计细胞来源和去除重复扩增序列。准确的细胞标记和UMI,就像为每个“乘客”核实身份,避免“冒名顶替”现象。在 cellranger count 运行过程中,Cell Ranger会识别R1序列中的Cell barcode和UMI,自动完成纠错、过滤低质量barcode和UMI等操作。
4. Reads比对与基因表达量统计(Alignment & Quantification)
使用比对工具(如STAR)将清洗后的序列准确定位到参考基因组上,并统计每个基因的表达量。最终产生每个细胞对应每个基因的表达矩阵。让每个片段回到基因的家,统计好表达量数据,为后续分析做好准备。Cell Ranger 使用 STAR 进行高效比对,将reads定位到参考基因组/转录组。
【通用模板】BAM转FASTQ(适用单细胞数据)
#安装samtools
conda install -c bioconda samtools
#转换BAM为FASTQ
# 单端
samtools fastq sample.bam > sample.fastq
# 双端(pair-end,适用于绝大多数单细胞,特别是10X/Smart-seq2)
samtools fastq -1 sample_R1.fastq -2 sample_R2.fastq -s sample_unpaired.fastq sample.bam
#-1 sample_R1.fastq :输出Read1
#-2 sample_R2.fastq :输出Read2
#-s sample_unpaired.fastq :输出未配对reads(一般很少用)
#sample.bam :你的输入文件, 若要加快速度可以用-@ 8指定8线程
#批量转换目录下所有bam
for bam in *.bam
do
base=$(basename $bam .bam)
samtools fastq -1 ${base}_R1.fastq -2 ${base}_R2.fastq -s ${base}_unpaired.fastq $bam
done
#转换得到的fastq文件,可直接作为cellranger、STARsolo等单细胞分析工具的输入。
#如果你的平台是Smart-seq2,R1和R2分别为转录本两端序列;如果是10X Genomics,R1往往包含barcode/UMI,R2是转录本序列,建议命名时注明R1/R2用途。【通用模板】BCL转FASTQ(适用单细胞数据)
# 安装bcl2fastq (仅需测序公司权限/技术)
conda install -c bioconda bcl2fastq
# 转换(举例)
bcl2fastq -R /path/to/BCL_folder -o /output/fastq_dir --sample-sheet /path/to/SampleSheet.csv
#这一步通常由测序公司完成。3. 实践中原始数据清洗方法与工具对比
3.1 比对与映射算法/参考序列类型对比
为什么要比对与选择参考序列?
单细胞测序数据的原始reads无法直接反映基因表达,需要通过比对(alignment/mapping)找到它们在基因组或转录组上的位置。比对的准确性直接决定了后续表达矩阵的质量。与此同时,所用的参考序列类型(如全基因组、已注释转录组、增强转录组)会影响我们能识别的基因种类、剪接体等信息。因此,合适的比对算法和参考选择对于保证数据解读的准确性、完整性具有基础性作用。
比对算法通常需要在速度、资源占用和信息保真度之间权衡;而参考序列的选择会影响到是否能够识别新型转录本或仅关注已知基因。因此,针对项目目标(如发现新转录本、分析已知基因表达等)进行合理选择,是单细胞分析流程中非常重要的环节。
映射算法比较
| Algorithm | 特点 | 应用场景/优缺点 |
|---|---|---|
| Spliced alignment | 能识别外显子-内含子拼接点,适用于RNA-seq | 适合检测新转录本,计算资源需求高(如STAR, HISAT2) |
| Contiguous alignment | 只对连续序列比对,忽略剪接信息 | 适用于无剪接/拼接简单的物种,适用性有限 |
| Lightweight mapping | 基于伪比对、kmer算法,速度极快,资源占用低 | 适用于大规模数据初筛,如Salmon, kallisto |
参考序列类型比较
| 参考类型 | 内容/结构特点 | 应用场景/优缺点 |
|---|---|---|
| Full reference genome | 完整基因组+注释 | 可发现新转录本/外显子,但比对慢、复杂 |
| Annotated transcriptome | 只含已注释的转录本 | 高效但遗漏新剪接体和新转录本 |
| Augmented transcriptome | 基于现有转录本加入新预测片段 | 平衡效率和信息丰富度,适合兼顾新旧信息需求 |
3.2 主流的单细胞数据处理工具包括:
-
Cell Ranger(10X官方推荐):集成barcode/UMI处理、序列比对和表达量定量,使用便捷,但计算量稍大。就像苹果设备用自家生态,简单高效,10X数据首选
-
STARsolo:基于STAR比对软件开发,快速、准确,且支持多种单细胞平台数据,适合大数据量。比喻为“特斯拉”,快速精准,效率高且灵活。
-
kallisto|bustools和Salmon alevin:通过伪比对算法提高速度,非常适合超大规模数据快速探索分析。就像“轻便型赛车”,轻量高效,适合超大数据量快速初筛。
| 工具路线 | 运算效率 | 适用数据类型 | 准确性 | 场景推荐 |
|---|---|---|---|---|
| Cell Ranger | 中等 | 10X数据专用 | 高 | 常规分析、标准流程 |
| STARsolo | 高 | 广泛适用 | 极高 | 中高数据量精准分析 |
| kallisto | 极高 | 广泛适用 | 中高 | 超大规模数据快速探索 |
| Salmon | 极高 | 广泛适用 | 中高 | 海量数据快速预处理 |
3.3 所需软件和环境配置
建议新手采用Conda或Docker进行环境管理,并能连接服务器运行,原始文件一般电脑跑不动,就像使用“便捷式厨房”,随时随地都能复现和共享分析环境。
标准代码示例(Cell Ranger):
# --------------------------
# 上传本地 fastq 文件夹到服务器
# 替换 username_local、server_ip、local_fastq_dir
# --------------------------
scp -r "/path/to/local_fastq_dir" username_local@server_ip:~
# 连接服务器(远程登录)
# 输入密码后即可登录
ssh username_local@server_ip
# 输入服务器密码
# --------------------------
# 激活分析环境(如 conda 环境),根据需要修改环境名
source ~/.bashrc
conda activate cellranger
# --------------------------
# 配置 cellranger 路径
# 替换为实际 cellranger 安装目录
export PATH=$PATH:/path/to/username/cellranger-8.0.1/
# --------------------------
# 进入数据目录,列出文件,进入目标文件夹
# 替换 filename 为实际文件夹名
ls
cd filename
# --------------------------
# 合并同一样本的不同 lane/批次的 fastq 文件为一个(以 read1 和 read2 为例)
# 替换 sample1_lane1_R1.fq.gz 等为实际文件名
# 可根据实际数量调整文件列表
cat sample1_lane1_R1.fq.gz sample1_lane2_R1.fq.gz sample1_lane3_R1.fq.gz undetermined_R1.fq.gz > sample1_merged_R1.fq.gz
cat sample1_lane1_R2.fq.gz sample1_lane2_R2.fq.gz sample1_lane3_R2.fq.gz undetermined_R2.fq.gz > sample1_merged_R2.fq.gz
# --------------------------
# 合并完成后重命名文件为标准命名
# 替换为你实际需要的命名规则
mv sample1_merged_R1.fq.gz projectSample_R1.fastq.gz
mv sample1_merged_R2.fq.gz projectSample_R2.fastq.gz
# --------------------------
# 运行 cellranger count 分析
# --id:输出文件夹名
# --transcriptome:参考转录组路径(改成你实际的参考文件夹)
# --create-bam=true:是否输出 bam 文件
# --fastqs:fastq 文件夹路径
# --sample:样本名(与 fastq 文件名一致)
cellranger count --id=output_folder_name \
--transcriptome=/path/to/username/reference_genome \
--create-bam=true \
--fastqs=/path/to/username/fastq_dir \
--sample=sample_name
# --------------------------
# 分析后下载结果文件夹回本地
# 替换 output_folder_name 和本地目标路径
scp -r username_local@server_ip:/path/to/username/fastq_dir/output_folder_name /path/to/local_save_dir
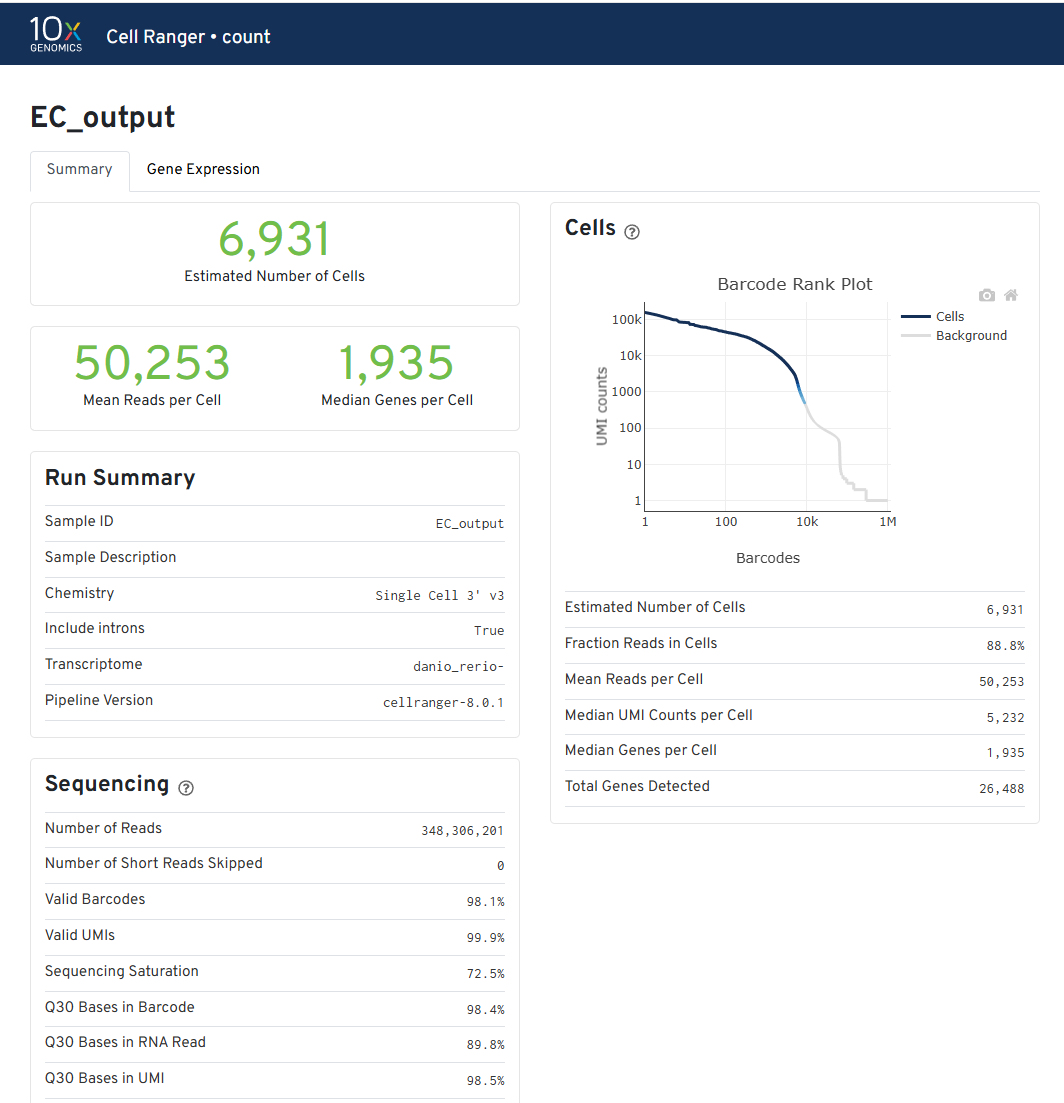
Fig. 3.4 . QC Report generated by Cell Ranger for single-cell RNA-seq data
标准代码示例(STARsolo):
STAR --genomeDir genome_index \
--readFilesIn read1.fastq read2.fastq \
--runThreadN 8 \
--soloType CB_UMI_Simple \
--soloCBwhitelist 10X_whitelist.txt
3.4 条形码(barcode)错误类型与解决方案
为什么会出现barcode与UMI相关问题?这些问题的危害是什么?
单细胞测序通过barcode和UMI技术,实现了数千上万细胞的高通量测序。但在细胞捕获、扩增和测序的实际流程中,条形码和UMI极易受到技术误差、液滴捕获偏差、PCR扩增和测序错误等因素影响。这些问题会引入虚假细胞(如空液滴)、混淆细胞身份(如双重/多重条形码),以及分子重复计数不准确,最终造成表达矩阵中细胞数异常、基因表达失真、假阳性/假阴性细胞类型等问题,严重影响后续聚类、注释和生物学解释。因此,在数据预处理阶段,及时识别和矫正这些barcode与UMI误差,是保障单细胞数据质量的关键。
-
双重/多重(Multiplet/Droplet doublet):一个barcode对应多个细胞,导致细胞计数偏低,常用DoubletFinder、Scrublet等工具检测并过滤。
-
空液滴(Empty droplet):无细胞液滴被barcode标记(环境RNA),如DropletUtils(R包)可通过统计分布自动判别并剔除。
-
序列错误(Sequencing Error):PCR或测序中误差导致barcode错配,导致同一细胞被拆分成多个barcode。通常用Levenshtein距离、whitelist纠正工具(如umi_tools, Cell Ranger自带模块)进行聚合和修正。
3.5 UMI resolution与矫正
UMI为什么需要纠正?
UMI是用于区分独立分子的短序列标签,理论上每个UMI对应一个原始RNA分子。但在实际扩增和测序过程中,UMI本身也可能发生测序错误或PCR突变,使原本属于同一分子的UMI被分为多个不同标签。这会导致重复计数,造成分子数被高估或低估。因此,对UMI进行错误矫正与归一化,是获得真实表达量、避免生物学假象的基础步骤。
UMI(Unique Molecular Identifier)是在建库时引入的短序列标签,用于标记每个分子。它用于去除PCR重复,准确估算原始分子的数量。
-
矫正步骤:
-
统计每个barcode下各UMI出现次数
-
合并相似UMI(如只差一个碱基的),以消除扩增误差
-
剔除仅出现一次、可能为测序噪音的UMI
-
常用工具如umi_tools、Cell Ranger内置模块均可实现UMI聚合和纠正。
4. 表达矩阵格式及后续分析步骤
完成数据清洗,我们终于得到期待已久的表达矩阵,它如同“基因表达的账单”,记录着每个细胞的基因活动状况。
4.1 表达矩阵的常见格式
-
稀疏矩阵(mtx):轻量、高效、节约内存。
-
Seurat对象(R):单细胞分析最主流的对象格式。
-
AnnData对象(Python):Scanpy生态的标准格式。
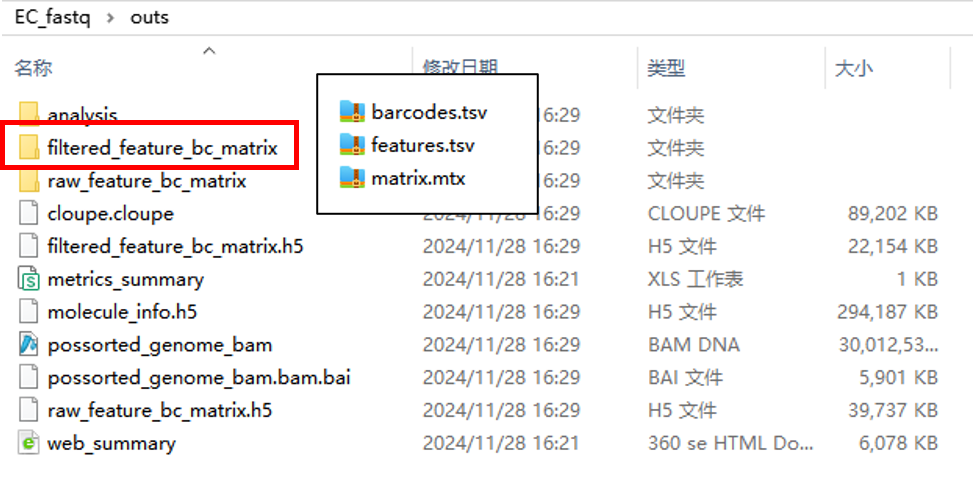
4.2 后续处理流程
-
基础质控(细胞/基因过滤、线粒体比例控制),去除“假细胞”和质量差的细胞,如“体检不合格”的数据,生成计数矩阵后,需全局评估数据质量,常用指标有:
-
Mapping rate:有效比对reads占总reads比例,过低提示样本问题。
-
每细胞UMI分布:反映细胞捕获/扩增效率。
-
每细胞基因数:低于阈值细胞常为“空液滴”或低质量,需过滤。
-
UMI去重率:高去重率说明PCR重复多,信息量有限。
-
线粒体基因比例:高比例提示细胞膜破损或死亡。
📝互动与交流
在单细胞数据清洗与处理的实际工作中,常常会遇到各种判断和选择。以下三个场景题,欢迎你留言讨论自己的选择理由,也可以思考自己的数据是否遇到过类似情况:
1. 你正在处理10X Genomics平台的单细胞RNA测序数据,发现有一部分细胞的线粒体基因表达比例异常高(大于20%)。这种现象最常见的原因是什么?在后续分析中你应采取什么措施?
A. 细胞活性很高,可以全部保留
B. 这些细胞可能受损或凋亡,应在质控步骤中过滤
C. 说明数据质量很好,可以直接下游分析
D. 说明样品污染,需要重新测序
2. 下列关于barcode和UMI的说法,哪一项是正确的?
A. barcode用于区分不同实验批次,UMI用于区分不同测序平台
B. barcode是在建库时加到每个细胞上的分子标签,UMI用于标记每个转录本的唯一身份
C. barcode和UMI都是分析结果后期计算出来的
D. UMI的数值越大,代表细胞质量越差
3. 你需要对一组Smart-seq2单细胞RNA测序数据进行分析,选择处理流程时应该注意什么?
A. 直接用Cell Ranger标准流程即可
B. Smart-seq2数据以每个细胞单独建库为主,推荐使用STAR、kallisto等全长转录本比对工具
C. 只需要去除低质量序列,无需比对
D. Smart-seq2不支持差异分析
欢迎在评论区留言你的答案和思考理由!你在实际项目中遇到过哪些数据清洗和质控的难题?你会如何判断和选择呢?说不定你的经验能帮到其他同路人!
下一期,我们将更进一步探讨单细胞表达矩阵的高级分析与可视化,敬请期待!

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐



 50
50 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)