Nphos:蛋白质 N-磷酸化数据库和预测器
摘要蛋白质 N-磷酸化广泛存在于自然界中,并参与各种生物过程。然而,与 O 型磷酸化相比,目前有关 N 型磷酸化的知识极为有限。在这项研究中,我们从 39 个物种的 7344 个蛋白质中收集了 11,710 个经实验验证的 N-磷酸化位点,随后构建了 Nphos 数据库,以分享蛋白质 N-磷酸化的最新信息。在这些大量数据的基础上,我们描述了蛋白质 N-磷酸化的顺序和结构特征。此外,在比较了数百个学
摘要
蛋白质 N-磷酸化广泛存在于自然界中,并参与各种生物过程。然而,与 O 型磷酸化相比,目前有关 N 型磷酸化的知识极为有限。在这项研究中,我们从 39 个物种的 7344 个蛋白质中收集了 11,710 个经实验验证的 N-磷酸化位点,随后构建了 Nphos 数据库,以分享蛋白质 N-磷酸化的最新信息。在这些大量数据的基础上,我们描述了蛋白质 N-磷酸化的顺序和结构特征。此外,在比较了数百个学习模型后,我们选择并优化了梯度提升决策树(GBDT)模型来预测人类的三种N-磷酸化类型,pHis、pLys和pArg的接收操作特征曲线下的平均面积(AUC)值分别为90.56%、91.24%和92.01%。同时,我们在人类蛋白质组中发现了 488,825 个不同的 N-磷酸位点。这些模型还被部署在 Nphos 中,用于交互式 N-磷酸复合预测。总之,这项工作为灵活而有针对性地研究 N-磷酸化提供了新的见解和要点。通过提供数据和技术基础,它还将有助于更深入、更系统地了解蛋白质的 N-磷酸化修饰。Nphos 可在 http://www.bio-add.org/Nphos/ 和 http://ppodd.org.cn/Nphos/ 免费获取。
引言
蛋白质 N-磷酸化是蛋白质磷酸化的一种自然形式[1],磷酸基团攻击精氨酸胍(pArg)、赖氨酸氨基(pLys)和组氨酸咪唑氮(pHis)形成磷酰胺键。越来越多的证据证实,蛋白质 N-磷酸化通过原核生物中的双组分信号转导,在中枢代谢[2]、趋化调节[3]、有氧/无氧调节[4]、孢子发生[5]和细胞分化[6]等方面发挥着关键作用[7]。最近,人们发现蛋白质 N-磷酸化与癌症进展有关 [8-10]。然而,与 O 磷酸化(磷酸丝氨酸、磷酸苏氨酸和磷酸酪氨酸)研究相比,由于 N 磷酸化的不稳定性[1]和检测方法的不先进,目前对 N 磷酸化的研究仍然停滞不前。
通常,低通量 32P 标记法[11] 和高通量质谱法(MS)[12] 被用来检测蛋白质的 N-磷酸化。然而,这两种方法都费力、费时,有时还不稳定。近年来,特异性抗体[13,14]和多肽富集方法[15-19]的引入大大提高了蛋白质 N-磷酸化检测的可靠性和效率。目前,与蛋白质磷酸化相关的数据库已超过 60 个[20],但只有 UniProt、PhosphoSitePlus[21]、dbPTM[22]、iPTMnet[23]和 dbPSP[24]等几个数据库除提供 O-磷酸化位点信息外,还提供零星的 N-磷酸化位点信息。在这些数据库中,PhosphoSitePlus、dbPTM 和 iPTMnet 是研究人员广泛使用的多功能综合数据库。除了提供磷酸化位点信息外,PhosphoSitePlus 还提供上游和下游激酶、磷酸酶和抗体的信息,以及磷酸化位点的生物过程。除了收集蛋白质磷酸化信息外,dbPTM 还包括 70 多种其他类型的翻译后修饰 (PTM) 信息。iPTMnet 数据库包含各种 PTM 的信息。它依赖于蛋白质信息资源(PIR)(https://protei ninformationresource.org/)。最近推出的 HisPhosSite 收集了 554 个实验验证的 pHis 位点和 15,378 个预测的 pHis 位点[25],是迄今为止唯一一个提供 N-磷酸化信息的数据库。不过,HisPhosSite 并不提供 pArg 和 pLys 位点的信息。
根据我们的深入调查,专门为预测 N-磷酸化位点而开发的硅学工具很少。大多数工具都是为 O 磷酸化设计的,而 O 磷酸化大多是激酶特异性的 [20,26,27],因此它们不适用于 N 磷酸化位点预测,因为目前只发现了少数 N 磷酸化特异性激酶。例如,McsB 是在枯草杆菌中发现的第一个针对蛋白质 pArg 位点的催化激酶 [28]。随后,在哺乳动物中发现了两种组氨酸蛋白激酶(NME1 和 NME2)和几种组氨酸蛋白磷酸酶(如 PHPT1 和 LHPP)以及少量底物[29]。近年来,pHisPred[30]、PROSPECT[31]和 iPhosH-PseAAC [32]三个模型分别基于 487、242 和 602 个 pHis 位点进行了大规模的 pHis 位点预测训练。然而,由于真核生物和原核生物可能不具有共同的 N-磷酸化机制,因此这些模型在真核生物 pHis 预测中的应用还值得商榷。毫无疑问,在实验方法取得技术突破之前,计算解决方案将成为蛋白质 N-磷酸化大规模研究的适用、经济、高效的解决方案。
为此,我们在本研究中从公共资源中收集了大量经过实验验证的 N-磷酸化位点。基于这些数据,我们构建了机器学习模型,用于全人类蛋白质组的蛋白质 N-磷酸化预测。最后,我们建立了一个新的数据库,以共享蛋白质 N-磷酸化的全面信息。我们预计,这项工作将成为各种规模的蛋白质 N-磷酸化研究的首选来源。
材料与方法
数据收集
N-磷酸化修饰的收集
我们使用多个关键词,如 "pHis"、"pArg"、"pLys"、"蛋白质组氨酸磷酸化"、"蛋白质精氨酸磷酸化",搜索了 PubMed 和 bioRxiv、"蛋白质赖氨酸磷酸化"、"磷酸精氨酸"、"精氨酸磷酸化"、"赖氨酸磷酸化"、"非典型磷酸化"、"磷酸化赖氨酸"、"N-磷蛋白体 "和 "pHisphorylation"。检索相关文章,并人工检查与 N-磷酸化的相关性。截至 2021 年 11 月,最终共检索到 115 篇文章。根据文章指南,从蛋白质组交换联盟(ProteomeXchange Consortium)(http://www.proteomexchange. org/)[36]下载了原始质谱数据,以便随后进行光谱解扰和数据分析。表 S4 列出了所有原始 MS 数据的详细信息。此外,我们还通过在 "MOD_RES "字段中使用 "phosphohistidine"、"phospholysine "或 "phosphoarginine "等关键词进行搜索,从 UniProt(截至 2020 年 6 月)中提取了 N-磷酸位点。只收集 PubMed 中有文献支持的条目。
蛋白质组数据处理
使用蛋白质组发现者(Proteome Discoverer)软件(v2.4;默认参数)对原始质谱文件进行处理,通过搜索UniProt Swiss-Prot Proteomes(2020年8月)的正向/反向组合数据库对肽段进行注释,该数据库涵盖智人(Homo sapiens)、丹顶鹤(Danio rerio)、枯草杆菌(B. subtilis)、金黄色葡萄球菌(Staphylococcus aureus)和大肠杆菌(E. coli)等多个物种。参数设置如下:酶,胰蛋白酶;前体质量容限,20 ppm;片段质量容限,0.6 Da(离子阱)或 0.02Da(轨道阱);光谱匹配,b、c、y、z 离子[电子转移/高能碰撞解离(EThcD)],b、y 离子[碰撞诱导解离(CID)或高能碰撞解离(HCD)],c、z 离子[电子转移解离(ETD)];动态修饰,Phospho/þ79.966 Da(H、K、R、S、T、Y);假发现率(FDR)目标值,0.05。使用 IMP-ptmRS 模块检测磷酸化肽,参数设置为默认值。所有 N-磷酸化数据均经过人工检查,并对重复的 N 磷酸化位点进行合并,以消除冗余和模糊性。
背景数据集
我们还从 UniProt 中的上述 6 个物种的所有蛋白质(表 S4)中提取了非磷酸化的 His、Arg 和 Lys 残基。这些非磷酸化位点被作为背景数据集,以消除 N-磷酸化可能造成的分类群偏差。
其他 PTMs 数据
除 N-磷酸化外,还从 PhosphoSitePlus [21]、iPTMnet [23] 和 dbPTM [33] 中获得了 O-磷酸化、甲基化、乙酰化和泛素化等多种 PTMs 的综合信息。对这些 PTM 位点进行了整合,并参照 UniProt 蛋白序列重新调整了它们的序列坐标。归一化的 PTM 数据被用作表征 N-磷酸化的参考数据集。
N 磷酸化位点的特征
磷酸化位点富集分析
E-比率的计算方法如下:
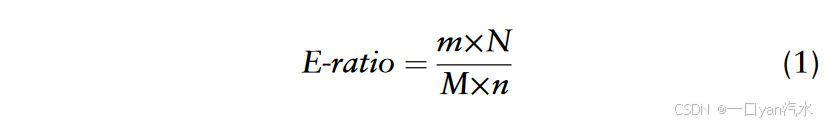
其中,m 是特定类型的磷酸位点的数量,n 是特定类型的非磷酸位点的数量,N 是所有非磷酸位点的数量,M 是所有磷酸位点的数量。磷酸化位点与非磷酸化位点之间富集度的比较使用 R 4.0.3 中的卡方检验法进行。
功能富集分析 GO 富集分析是使用 R 软件包 "clusterProfiler"(pvalueCutoff 1⁄4 0.05,pAdjustMethod 1⁄4 "BH";BH 指 Benjamini and Hochberg),以所有选定物种的 N-磷酸位点的 UniProt GO 条目为背景(背景数据集)进行的。蛋白质类别分析采用 PANTHER 15 (http://pantherdb.org/) [37]。
序列和结构模式分析
使用 R 软件包 "rmotifx"[38] 提取 N-磷酸化位点周围潜在的序列模式(得分≥ 5,出现率≥ 10)。先前的一项研究表明,磷酸化特异性与磷酸化位点周围的主序列密切相关[39],其中磷酸化位点周围的上游/下游 14 个氨基酸是保守的[40]。因此,我们采用了一个以 31 个连续氨基酸为中心的磷酸化窗口[41]。此外,还使用 R 软件包 "ggseqlogo "生成序列标识,并根据残基的理化性质进行着色。整体序列模式通过在线服务 pLogo [42] 可视化(https://plogo.uconn.edu/)。上述背景数据集被用作主题分析的对照。
二级结构和表面可及性由 SPOT-1D-Single [43] 预测,采用默认参数。有序区和无序区由 IUPred3 [44] 评估,采用默认参数。
为构建模型准备数据集
阳性数据集
阳性数据集包括所有经实验验证的人类 N-磷酸位点。以磷酸化位点为中心残基,提取 31 个连续氨基酸的肽作为阳性肽。多余的肽被排除在外。阳性肽由 8978 个不同的 31 个残基肽组成,涵盖人类蛋白质组中 5798 个不同的蛋白质(图 5)。
阴性数据集
实际上,由于实验验证的 N-磷酸化位点(阳性位点)数量有限,因此确定非磷酸化仍是一个未决问题。作为替代方案,我们分别生成了非磷酸化 His、Lys 和 Arg 的阴性数据集,具体如下。(1) 挑选 N-磷酸化蛋白质中的非磷酸化 His/Lys/Arg 残基。(2) 排除暴露区域的残基,这些残基由 RSA 评估。RSA 阈值根据氨基酸的数据集平衡确定,即 His 的 RSA ≤ 0.12,Lys 的 RSA ≤ 0.3,Arg 的 RSA ≤ 0.2。(3) 生成阴性肽(以非磷酸化的 His、Lys 或 Arg 为中心的 31 个残基序列)。(4) 通过对阴性序列与阳性数据集进行同源性分析,去除错误或含混的阴性序列。同源性分析使用 CD-HIT [45],将序列同一性阈值设为 30%。表 4 列出了所得到的阴性数据集的信息。
特征转换
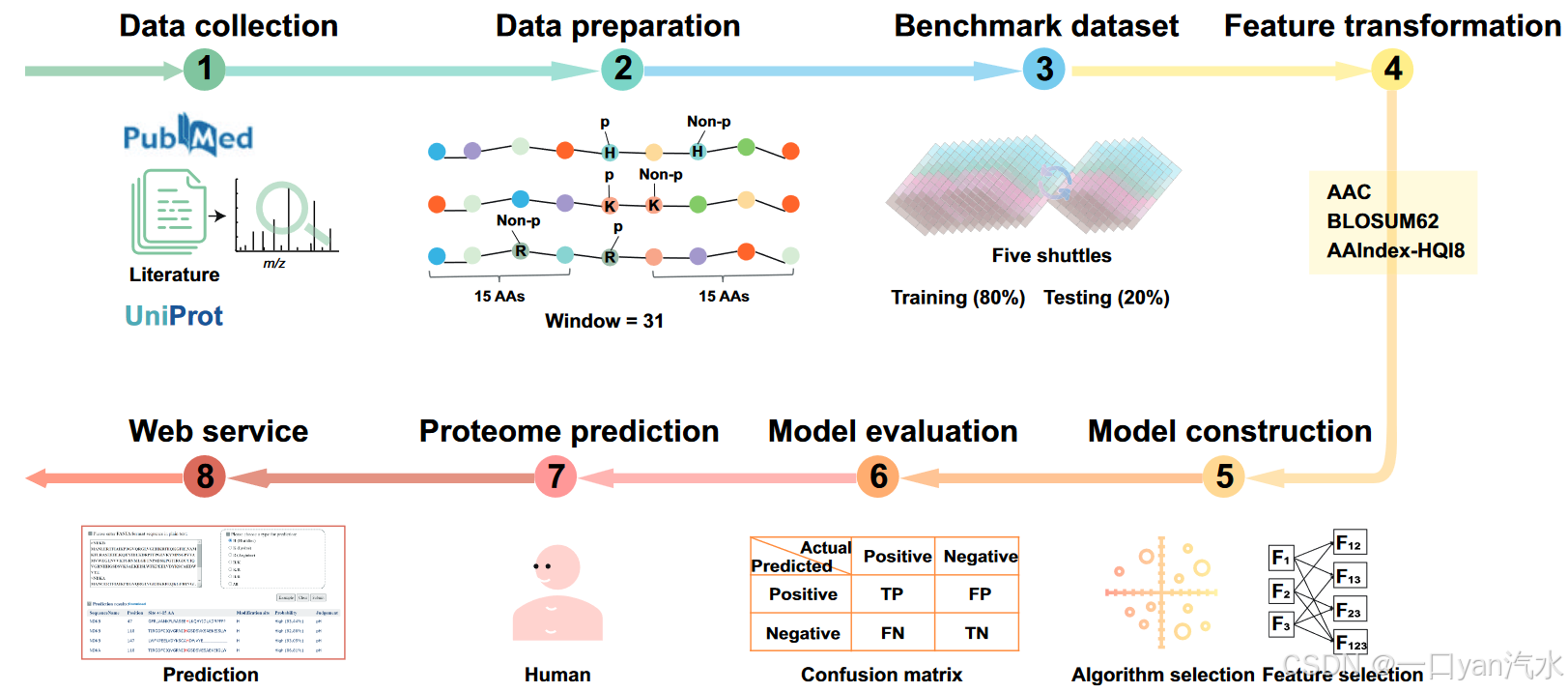
AAindex,氨基酸指数;TP,真阳性;FP,假阳性;FN,假阴性;TN,真阴性;Window,用于提取特征的序列窗口长度。
每个 31 位残基肽(阳性和阴性)在应用于模型构建之前都被转换成 1 × X 的特征向量(图 5)。特征向量使用三组不同特征的任意组合进行编码:AAC、氨基酸指数(AAindex)-HQI8 和 BLOSUM62。
氨基酸指数(AAC)表示肽中 20 个正常氨基酸的频率,计算公式如下:
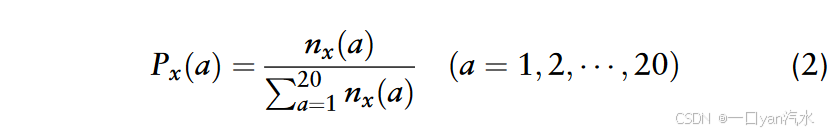
其中,(a) 表示氨基酸 x 的数量。
AAindex [46] (https://www.genome.jp/aaindex/) 是一个包含 20 种氨基酸的物理、化学和生物特性的数据库。目前,每种氨基酸有 566 种特性。为了克服属性数量过多带来的过拟合问题,Sahara 等人采用模糊聚类法对中心属性进行聚类和选举,从而开发出质量指标 -HQI8 [47]。在本研究中,我们也使用 HQI8 提取多肽的理化特征,并进一步将其转换为 1 × 248 特征向量。
BLOSUM62 矩阵是一个 20 × 21 维的氨基酸替换矩阵,通常用于确定两个肽序列的相似性。在该矩阵中,每个条目都是几率分数的对数,它是用氨基酸对出现的频率除以氨基酸随机排列的可能性得出的[48]。
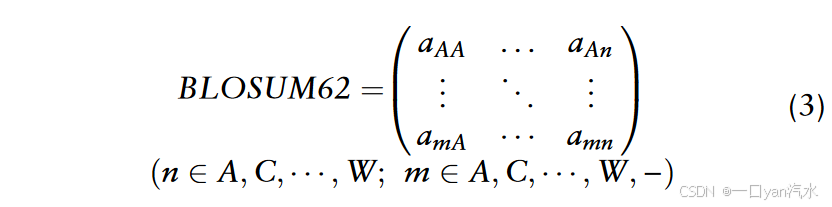
BLOSUM62 矩阵 的每一行都代表一个氨基酸与其他氨基酸的保守替换:
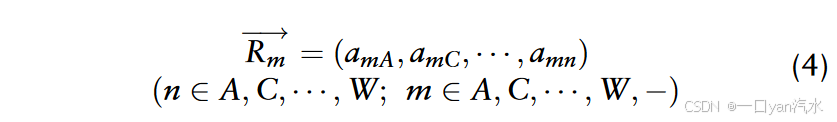
值得注意的是,BLOSUM62 矩阵由 20 个规范字符(对应 20 个氨基酸)和一个非规范字符("-",20 个氨基酸中的任意一个)组成。这样,31 个残基的多肽就被转化成了 1 × 620 的特征向量 ,如下:
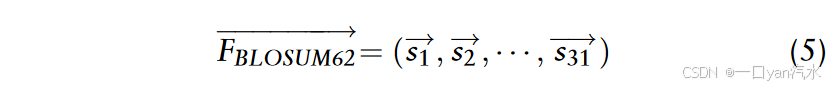
其中,s 代表 20 种氨基酸中的任意一种, (如果s=m,m属于A,C,...,W,-)
在本研究中,AAC、AAindex-HQI8 和 BLOSUM62 特征向量是通过自编码的 Python 脚本计算得出的。BLOSUM62 矩阵来自 Henikoff 和 Henikoff 的研究 [48]。这三个特征向量的组合是通过相互连接产生的。
构建模型的算法
分别针对 pHis、pLys 和 pArg 构建了 N-磷酸化预测的机器学习模型。模型构建总共采用了 10 种不同的算法,包括 AdaBoost、人工神经网络(ANN)、CatBoost、决策树(DT)、GBDT [49]、k-近邻(KNN)、逻辑回归(LR)、天真贝叶斯(NB)、二次判别分析(QDA)和随机森林(RF)。除 CatBoost 外,所有算法均通过 Python 软件包 scikit-learn (v1.0.2) 调用。CatBoost 算法通过 Python 包 catboost(v1.0.4)调用。表 S5 总结了调用方法。参数细化是通过 sklearn.model_selection.GridSearchCV (cvc) 进行的。GridSearchCV (cv =10).
模型训练和性能评估
在本研究中,为了确定 N-磷酸化预测的全局最优条件,我们基于七种特征向量组合和十种学习算法进行了全面的组合学习(图 5)。因此,我们为三种 N-磷酸化类型(pHis、pLys 和 pArg)分别构建并比较了 70 个不同的模型。表 S5 总结了每种机器学习算法的相应最优超参数(如适用)。
在构建每个模型时,将正反面随机分成两部分。具体来说,80% 用于模型训练和内部评估,其余 20% 用于模型验证(图 5)。为了增强模型的泛化能力,同样的操作重复了五次,取平均值作为模型的最终性能。同时,为了巩固稳健性,还通过 10 倍交叉验证进行了内部评估,将正负数据随机分成 10 倍,其中 9 倍用于构建模型,1 倍用于内部评估。10 倍交叉验证重复了 10 次。
模型性能由多个参数评估,包括灵敏度或召回率、特异性、精确度、F1score、准确度和 MCC,具体如下:

其中,TP、TN、FP 和 FN 分别代表真阳性、真阴性、假阳性和假阴性的数量。此外,还绘制了 ROC 曲线,并确定了 AUC。
数据库构建
数据库是在 Linux þ Tomcat þ Java 的系统架构上构建的。采用 MySQL 作为底层数据库管理系统。网页采用 HTML5 技术编码,支持移动访问。我们在多种互联网浏览器上测试了在线服务,包括 Mozilla Firefox、Google Chrome 和 Microsoft Edge。
结果
人类 N-磷酸化的大规模预测
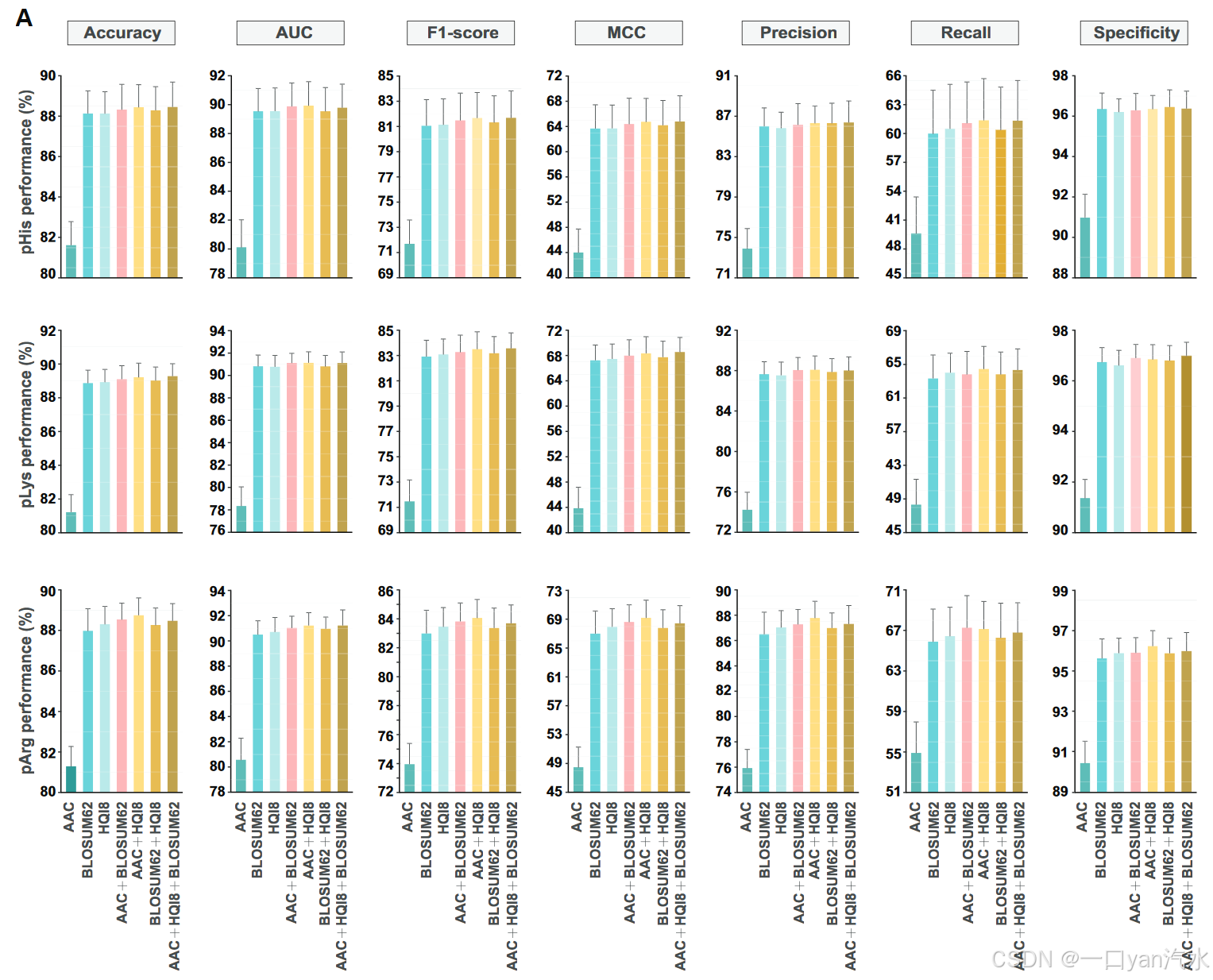
算法和特征向量的选择
为了选择算法和特征向量的最佳组合,我们针对每种 N-磷酸化类型分别构建了 70 个不同的模型。我们对模型的性能进行了全面评估和比较,最终确定了最佳组合。梯度提升决策树(GBDT)算法在所有三种 N-磷酸化类型的预测中均优于其他算法。在特征向量方面,pHis 模型偏好氨基酸组成(AAC)+ 高质量指数(HQI8),pLys 模型偏好 AAC + HQI8 + BLOSUM62,pArg 模型偏好 AAC + HQI8(图 3A;表 S1)。在预测 N-磷酸化时,特征向量具有不同偏好的确切原因仍不清楚。因此,我们重新构建了用于 N-磷酸化预测的 GBDT 模型,并重复了超参数优化以获得最佳性能(表 S2)。
GBDT分类器的性能评估
基于五个随机洗牌训练数据集的GBDT模型的十倍交叉验证巩固了N-磷酸化预测的良好性能,在预测pHis、pLys和pArg时,接收者操作特征曲线(ROC)下的平均面积(AUC)值分别为90.65%、91.54%和91.78%(表S3)。使用五个随机洗牌测试数据集进一步证实了模型的良好性能,预测 pHis、pLys 和 pArg 的平均 AUC 值分别为 90.56%、91.24% 和 92.01%(表 1)。
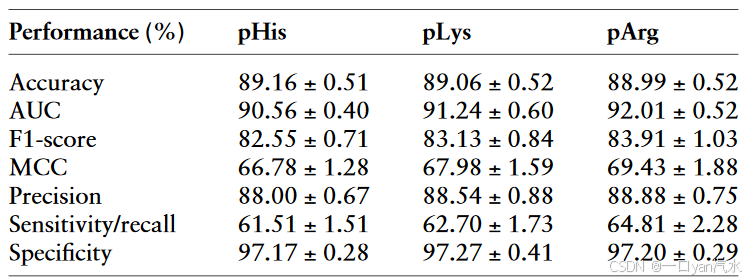
注:pHis、pLys 和 pArg 分别表示组氨酸、赖氨酸和精氨酸的磷酸化。AUC,ROC 曲线下面积;ROC,接收者操作特征;MCC,马修斯相关系数;GBDT,梯度提升决策树。
此外,我们还检查了错误预测的数据。在所有错误预测事件中,81.08%为假阴性(pHis、pLys 和 pArg 分别为 19.32%、37.06% 和 24.70%)。与所有阳性和真阳性相比,很大比例的假阴性位于非暴露、非卷曲或有序区域(图 3B),这表明它们是次要的阳性类型。对这些假阴性的进一步序列模式分析也未能发现与实验验证的磷酸位点(所有阳性)一致的序列特征(图 2A 和 3C)。因此,我们推测这些阳性结果在模型学习过程中代表性不足。在模型训练中加入更多类型的阳性将大大提高模型的性能。
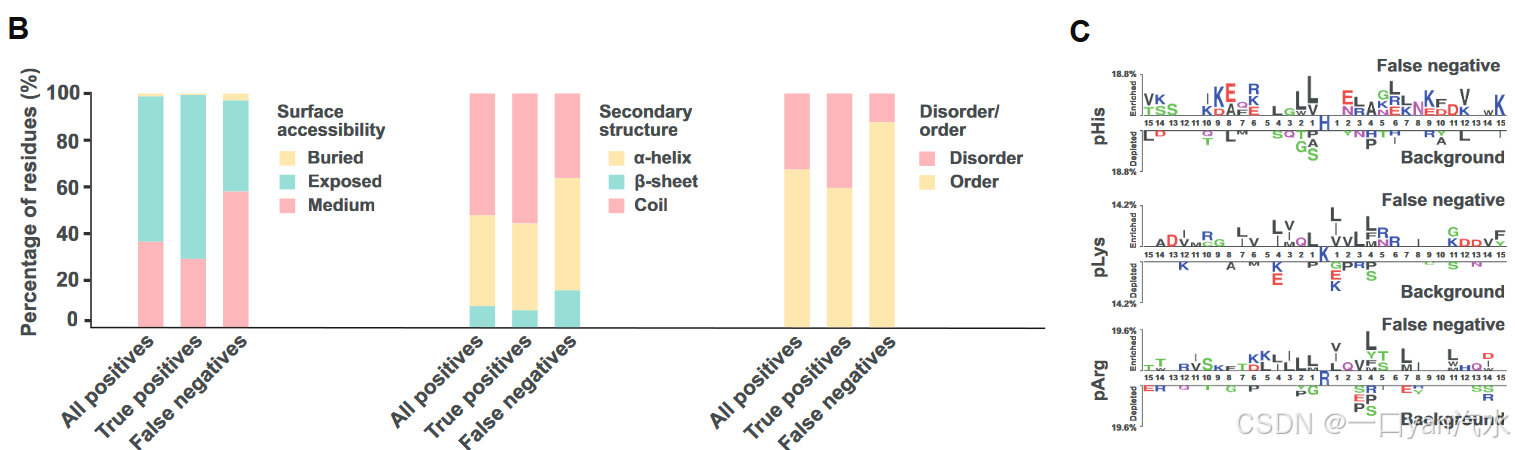
C. 假阴性的序列模式。AAC,氨基酸组成;HQI8,高质量指数;MCC,马修斯相关系数;AUC,ROC 曲线下面积;ROC,接收者操作特征;GBDT,梯度提升决策树。
与其他 pHis 预测器的比较
以前,有三种预测器,即 iPhosH-PseAAC [32]、PROSPECT [31] 和 pHisPred [30],被用来预测蛋白质的 pHis 位点。PROSPECT 是大肠杆菌特异性 pHis 预测器;iPhosH-PseAAC 是一般 pHis 预测器;pHisPred 是真核细胞特异性和原核细胞特异性 pHis 预测器;Nphos 是人类特异性 pHis/pLys/pArg 预测器(表 2)。Nphos 的马修斯相关系数(MCC)和 F1 分数总体上优于这些预测因子。虽然 iPhosH-PseAAC 采用了深度学习算法(更准确地说,是多层反向传播神经),取得了显著的召回值(检测阳性的灵敏度)和 MCC,但其通用性和鲁棒性尚未得到适当评估。重要的是,与其他预测器相比,Nphos 是在一个更大(超过两倍)、更集中(只有人类 pHis)的阳性数据集上构建的,从而保证了更好的可靠性。正如我们所描述的那样,真核生物和原核生物的 N-磷酸化在顺序和结构方面都存在显著差异,将不同物种的所有 pHis 事件都纳入模型学习可能是早期预测方法的一个主要缺陷。除了所有预测因子中使用的生成阴性结果的排他性策略外,Nphos 还对非 His 和 pHis 的溶剂可及性提供了额外的限制(设置相对溶剂可及性(RSA)阈值),以进行大规模的磷酸化鉴定,从而确保阴性结果更具可解释性,更接近实际情况。
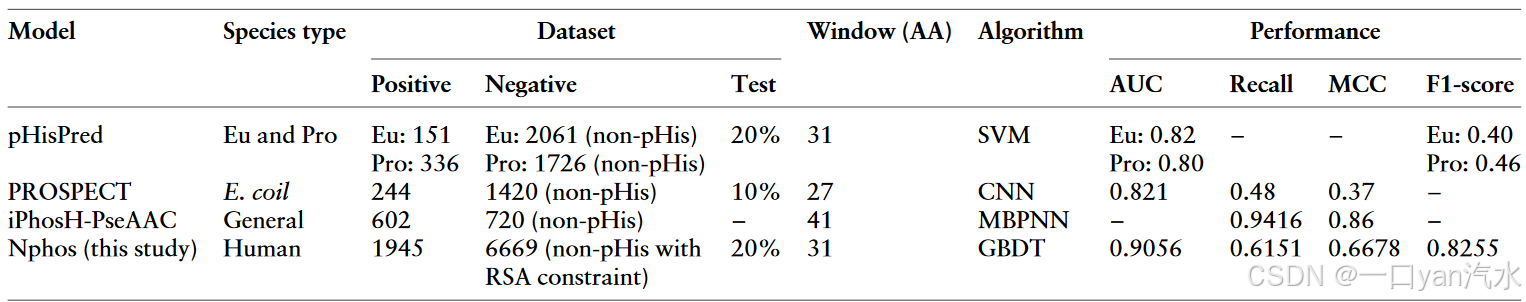
注:"Negative(阴性)"和 "Positive(阳性)"分别指阴性数据集和阳性数据集的数量;"Test(测试)"指测试数据集占总数据集的百分比;"Window(窗口)"表示用于提取特征的序列窗口的长度。AA,氨基酸;Eu,真核生物;Pro,原核生物;E. coil,大肠杆菌;CNN,卷积神经网络;MBPNN,多层反向传播神经;SVM,支持向量机;-,不可用。
全蛋白质组人类 N-磷酸化预测
利用训练有素的 GBDT 分类器,我们对人类蛋白质组中的 N-磷酸化进行了大规模预测。人类蛋白质组序列来自 UniProt 知识库(截至 2022 年 2 月 25 日)。为获得高度可靠的N-磷酸化位点,设定的概率阈值为85%。排除实验验证的位点,在 20,259 个人类蛋白质中预测出 488,825 个不同的 N-磷酸位点,包括 64,409 个 pHis(15,192 个蛋白质)、214,679 个 pLys(19,025 个蛋白质)和 209,737 个 pArg 位点(19,364 个蛋白质)。这使得 pHis、pLys 和 pArg 的 N-磷酸化修饰百分比与背景氨基酸用量相比分别增加到 21.61%、32.97% 和 32.75%(表 3)。这些数值与实验验证的人类 O 型磷酸化修饰值相当。重要的是,大约74.40%的实验验证的N-磷酸化蛋白和87.12%的预测的N-磷酸化蛋白也发生了O-磷酸化,这意味着O-磷酸化和N-磷酸化之间可能存在串扰。
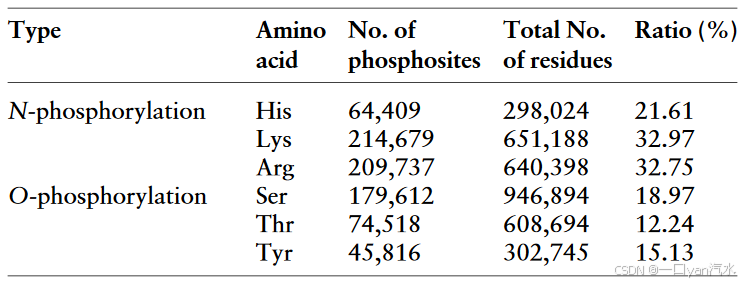
注:N-磷酸位点由 GBDT 模型预测,
而 O-磷酸位点则是经实验验证的位点,这些位点来自正文所述的多种来源。
讨论
自蛋白质 N-磷酸化首次被发现以来,已经过去了半个多世纪。遗憾的是,与蛋白质 O 磷酸化或其他 PTM(如泛素化、乙酰化、甲基化和 SUMOylation)相比,N 磷酸化的功能研究受到了稳定检测 N 磷酸化位点的技术难度的限制。然而,不断的努力提供了有关 N-磷酸化的宝贵数据。遗憾的是,这些数据在发表后仍然没有得到利用,而且很少有人尝试收集这些数据并重新利用它们。HisPhosSite [25] 是第一个蛋白质 N-磷酸化数据库,它包含 411 个蛋白质中 554 个经实验验证的 pHis 位点。这些位点已被纳入 Nphos。因此,本研究向前迈出了重要一步,引入了新型数据库 Nphos,该数据库全面收集了 39 个物种 7344 个蛋白质中 11,710 个经实验验证的 N-磷酸化位点,涵盖 pHis、pLys 和 pArg 位点。利用该数据库,我们进行了大规模分析,以确定 N-磷酸化修饰的顺序和结构特征。例如,我们发现 pLys 和 pArg 可能需要富含 SP 的基序,这是以前从未报道过的发现。
在这项研究中,我们优先使用机器学习算法,即基于 GBDT 的模型,来预测人类蛋白质组中的 pHis、pLys 和 pArg 位点,以广泛扩展 N-磷酸化的知识。这些模型还被部署为一项在线服务,用于从原始蛋白质序列中交互预测 N-磷酸化。这大大增强了对 N-磷酸化修饰进行大规模和重点功能研究的能力。
毫无疑问,本研究的分析和预测涉及到迄今为止经实验验证的 N-磷酸化位点,即使从公共资源中进行了彻底收集,这些位点仍然有限。N-磷酸化类型的代表性不足解释了模型预测灵敏度相对较低的原因。此外,底片的生成对于决策模型至关重要。遗憾的是,这项研究还没有完全解决这个问题。如果能获得更多经过实验验证的 N-磷酸化位点,并引入新的学习算法(如无阿贝尔学习法),将大大提高 N-磷酸化预测的准确性。此外,输入肽段的长度也会影响模型的性能。一些 N/O 磷酸化预测模型通过选择不同的肽段长度来优化模型性能[20]。例如,在 pHisPred 中,不同的机器学习算法对肽段长度有不同的敏感性 [30]。因此,未来有望选择优化的序列长度来构建模型。
未来发展
实际上,所有关于 N-磷酸化的研究都仅限于目前已确定的磷酸化蛋白。我们预计,未来可用的 N-磷酸化数据将大幅增长,这凸显了发现和更好地定义假定 N-磷酸化位点的重要性。未来,我们计划扩展 Nphos 中的可用数据,包括有关介导修饰的激酶和磷酸酶、蛋白质-蛋白质相互作用、串扰、结构域、疾病关系和结构可视化的信息。此外,我们还将开发用于嵌入式主题分析、批量搜索功能和进化分析的新工具。此外,最近深度学习算法在各种 PTM 位点预测任务中的应用[20,35]也启发我们充分利用前沿人工智能技术准确预测 N-磷酸化。
结论
这项研究提供了迄今为止有关蛋白质 N-磷酸化的最有价值的信息。模型预测将指导抗体的精确设计,以验证特定的 N-磷酸化事件并进一步探索其生物学功能。最后,这项工作将克服目前对灵活而集中的蛋白质 N-磷酸化研究的实验限制。特别是,通过研究 N-磷酸化和 O-磷酸化之间的关系,它将有助于发现激酶。

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐
 20
20 0
0- 0



 已为社区贡献3条内容
已为社区贡献3条内容

所有评论(0)