Graph图和Geometric Deep Learning几何深度学习的Foundation Models基础模型

本文约10000字,建议阅读10+分钟
本文为你介绍图 FM及其使用的示例。语言、视觉和音频的Foundation Models基础模型已成为 2024 年机器学习的主要研究主题之一,而针对图结构数据的 FMs 则有些落后。在这篇文章中,我们认为图 FM 的时代已经开始,并提供了一些如何在今天使用它们的示例。
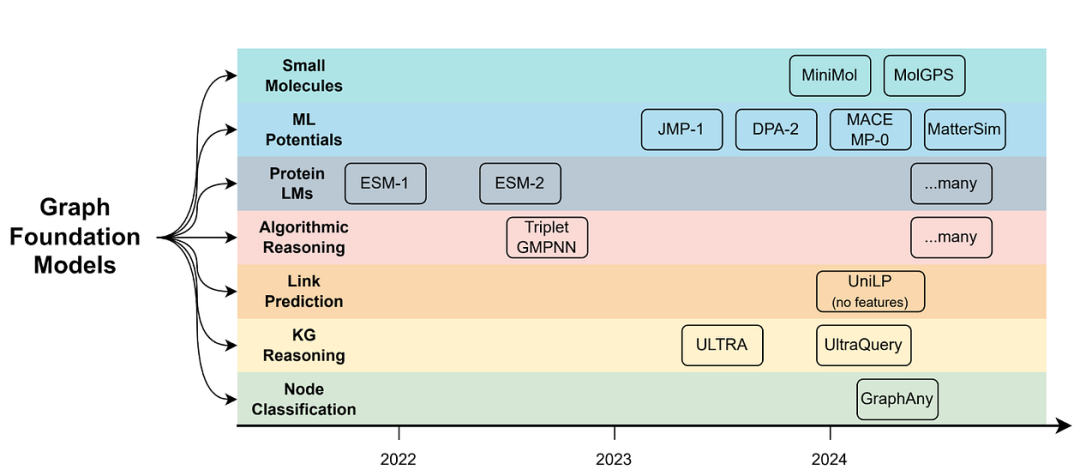
图形和几何深度学习中新兴基础模型的时间线。
什么是图形基础模型以及如何构建它们?
由于对于什么是“基础”模型存在一定程度的模糊性,因此首先进行定义以建立共同点是适当的:
“图形基础模型是一种单一(神经)模型,可以学习可转移的图形表示,并可以推广到任何新的、以前从未见过的图形”
其中一个挑战是图具有各种形式和形状,并且它们的连接和特征结构可能非常不同。标准图神经网络 (GNN) 不是“基础性的”,因为它们最好只能在具有相同类型和特征维度的图上工作。可以在任何图上运行的图启发式方法(例如标签传播https://en.wikipedia.org/wiki/Label_propagation_algorithm或个性化 PageRank https://en.wikipedia.org/wiki/PageRank)也不能被视为图 FM,因为它们不涉及任何学习。尽管我们喜欢大型语言模型,但目前仍不清楚将图解析为可以传递给 LLM 的序列(例如在GraphText https://arxiv.org/abs/2310.01089或Talk Like A Graph https://openreview.net/forum?id=IuXR1CCrSi中)是否是一种合适的方法,可以保留图对称性并扩展到比玩具大小的数据集更大的任何东西(我们将 LLM + 图留在单独的帖子中)。
设计 Graph FM 最重要的问题可能是可迁移的图形表示。正如Mao、Chen 等人在最近的 ICML 2024 立场文件https://arxiv.org/abs/2402.02216中所建议的那样,LLM 可以将任何语言中的任何文本压缩为固定大小词汇表中的标记。视频语言 FM 诉诸于始终可以从图像中提取的补丁(任何图像或视频中始终都有 RGB 通道)。目前尚不清楚对于可能具有非常多样化特征的图形,通用特征化(标记化)方案是什么,例如:
-
一个具有节点特征和一些给定节点标签的大图(典型用于节点分类任务)
-
一个没有节点特征和类别但具有有意义的边类型的大图(典型用于链接预测和 KG 推理)
-
许多具有/不具有节点/边特征的小图,具有图级标签(典型用于图分类和回归)
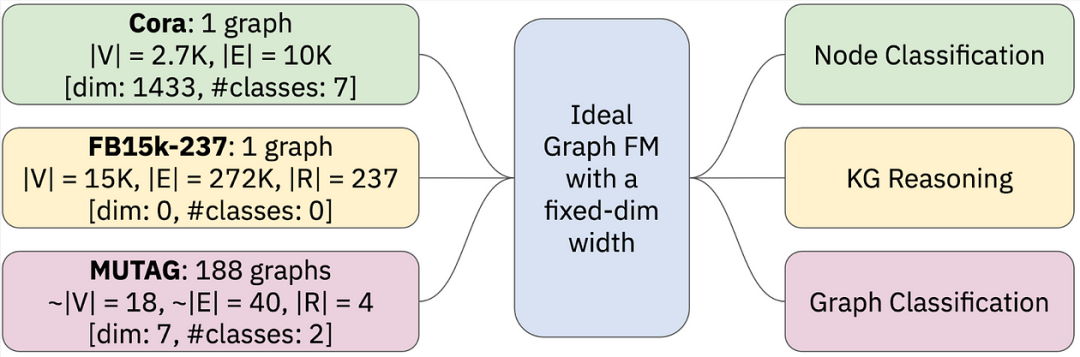
一个理想的图基础模型,它采用具有任何节点/边/图特征的任何图并执行任何节点/边/图级任务。截至 2024 年中期,此类图 FM 尚不存在纯粹形式。
到目前为止,图学习社区在设计图 FM 时还有几个未解决的研究问题:
1️⃣ 如何在具有异构节点/边/图特征的图中进行泛化?例如,用于节点分类的流行Cora数据集是一个具有维度 1,433 的节点特征的图,而 Citeseer 数据集具有 3,703 维特征。如何为如此多样化的图定义一个单一的表示空间?
2️⃣ 如何在预测任务中推广?节点分类任务可能具有不同数量的节点类别(例如,Cora 有 7 个类别,而 Citeseer 有 6 个)。更进一步,节点分类模型能否在链接预测中表现良好?
3️⃣ 基础模型的表达能力应该是什么?人们对 GNN 的表达能力进行了大量研究,通常采用与 Weisfeiler-Lehman 同构测试的类比。由于图基础模型理想情况下应该处理广泛的问题,因此正确的表达能力难以捉摸。例如,在节点分类任务中,节点特征与图同质性或异质性一样重要。在链接预测中,结构模式和打破自同构更为重要(节点特征通常不会带来巨大的性能提升)。在图级任务中,图同构开始发挥关键作用。在分子生成等 3D 几何任务中,需要处理连续对称性的额外复杂性(参见《几何 GNN 的漫游指南》https://arxiv.org/abs/2312.07511)。
在以下部分中,我们将展示至少在某些任务和领域中,Graph FM 已经可用。我们将重点介绍它们在可迁移特性方面的设计选择以及在对新的未见图进行归纳推理方面的实际优势。
📚更多内容请阅读参考文献[1][2]和Github Repo https://github.com/CurryTang/Awesome_Graph_Foundation_Models
节点分类:GraphAny
多年来,基于 GNN 的节点分类器一直局限于单个图数据集。也就是说,例如,给定具有 2.7K 个节点、1433 维特征和 7 个类的 Cora 图,必须专门在 Cora 图上训练一个带有其标签的 GNN,并在同一图上运行推理。将训练好的模型应用于另一个图(例如具有 3703 维特征和 6 个类的 Citeseer)将遇到难以克服的困难:一个模型如何推广到不同的输入特征维度和不同数量的类?通常,预测头被硬编码到固定数量的类。
据我们所知, GraphAny https://arxiv.org/abs/2405.20445是第一个 Graph FM,其中单个预训练模型可以对具有任何特征维度和任何数量类别的任何图执行节点分类。在标准威斯康星数据集https://pytorch-geometric.readthedocs.io/en/latest/generated/torch_geometric.datasets.WebKB.html#torch_geometric.datasets.WebKB的 120 个节点上进行预训练的单个 GraphAny 模型成功推广到 30 多个不同大小和特征的其他图,并且平均而言,其表现优于在每个图上从头训练的 GCN 和 GAT 图神经网络架构。

GraphAny 概述:LinearGNN 用于执行非参数预测并得出熵归一化的距离特征。最终预测是通过融合每个节点上的多个 LinearGNN 预测和基于距离特征学习的注意力来生成的。
设置:半监督节点分类:给定图 G、节点特征 X 和来自 C 类的几个标记节点,预测目标节点的标签(二分类或多分类)。节点特征的维度和唯一类的数量不是固定的,并且依赖于图。
可迁移性:GraphAny 不会为所有可能的图形建模一个通用的潜在空间(这非常麻烦,甚至可能实际上是不可能的),而是绕过这个问题,专注于光谱滤波器预测之间的相互作用。给定一组类似于简化图卷积https://arxiv.org/abs/1902.07153的高通和低通滤波器(例如,AX 和 (IA)X 形式的操作,在论文中称为“LinearGNN”)和已知节点标签:
0️⃣ GraphAny 将过滤器应用于所有节点;
1️⃣ GraphAny 通过求解封闭形式的最小二乘优化问题,从具有已知标签的节点中获得每个预测器的最佳权重(最佳权重表示为伪逆);
2️⃣ 将最佳权重应用于未知节点以获得暂定的预测日志;
3️⃣ 计算这些 logit 之间的成对距离并应用熵正则化(这样不同的图形和特征大小就不会影响分布)。例如,对于 5 个 LinearGNN,这将产生 5 x 4 = 20 个 logit 分数组合;
4️⃣ 学习这些逻辑上的归纳注意力矩阵,以最有效地对预测进行加权(例如,将更多注意力放在异质图的高通滤波器上)。
最后,模型中唯一可学习的组件是注意力的参数化(通过 MLP),它不依赖于唯一类的目标数量,而只依赖于使用的 LinearGNN 的数量。同样,所有 LinearGNN 预测器都是非参数的,它们的更新节点特征和最佳权重可以预先计算,以便更快地进行推理。
📚更多内容请阅读参考文献 [3]
链接预测:尚未
设置:给定一个图 G,有或没有节点特征,预测一对节点 (v1, v2) 之间是否存在链接
对于具有节点特征的图,我们不知道任何用于链接预测的单一可转移模型。
对于非特征化图(或者当你决定故意省略节点特征时),还有很多话要说——基本上,由于统一的节点特征化策略,所有具有标记技巧的 GNN 都可能转移到新的图。
众所周知,在链接预测中,最大的障碍是自同构节点(具有相同结构角色的节点)的存在——普通 GNN 为它们分配相同的特征,使得下图中的两个链接 (v1, v2) 和 (v1, v3) 难以区分。双半径节点标记https://proceedings.neurips.cc/paper/2018/hash/53f0d7c537d99b3824f0f99d62ea2428-Abstract.html或距离编码https://proceedings.neurips.cc/paper_files/paper/2020/hash/2f73168bf3656f697507752ec592c437-Abstract.html等标记技巧https://arxiv.org/abs/2010.16103是破坏自同构对称性的节点特征化策略。
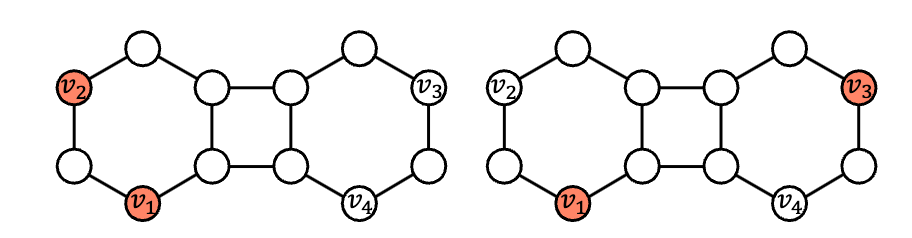
V2 和 v3 是自同构节点,标准 GNN 对 (v1,v2) 和 (v1,v3) 的评分相同。当我们预测 (v1, v2) 时,我们会将这两个节点标记为与其他节点不同,以便 GNN 在学习 v1 和 v2 的表示时能够意识到目标链接。同样,在预测 (v1, v3) 时,节点 v1 和 v3 将被标记为不同。这样,左图中 v2 的表示将与右图中 v3 的表示不同,从而使 GNN 能够区分非同构链接 (v1, v2) 和 (v1, v3)。
也许唯一一种使用标记技巧(针对非特征化图)并在未见图的链接预测上进行评估的方法是UniLP。UniLP https://arxiv.org/abs/2402.07738是一种上下文对比学习模型,需要为每个要预测的目标链接提供一组正样本和负样本。实际上,UniLP 使用SEAL作为骨干 GNN,并在固定数量的正样本和负样本上学习注意力。另一方面,SEAL https://proceedings.neurips.cc/paper/2018/hash/53f0d7c537d99b3824f0f99d62ea2428-Abstract.html 的速度非常慢,因此让 UniLP 扩展到大型图的第一步是用更有效的方法(如ELPH https://arxiv.org/abs/2209.15486和BUDDY https://arxiv.org/abs/2209.15486)取代子图挖掘。
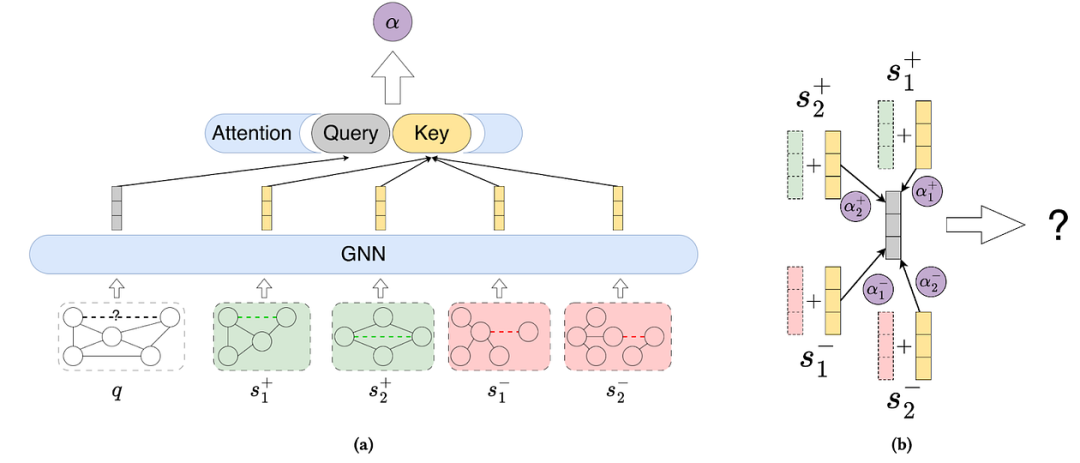
通用链接预测器框架概述。(a)为了预测查询链接𝑞,我们首先从目标图中采样正(𝑠+)和负(𝑠-)上下文链接。查询链接和这些上下文链接都通过共享子图 GNN 编码器独立处理。然后,注意力机制根据查询链接和上下文链接之间的相似性计算分数。(b)通过加权求和获得由目标图上下文化的查询链接的最终表示,该加权求和将上下文链接的表示与其各自的标签相结合。
可转移的:通过标记技巧 GNN 学习到的结构模式——事实证明,像Neural Bellman-Ford https://arxiv.org/abs/2106.06935这样的方法可以捕获节点对的指标,例如个性化 PageRank 或 Katz 索引(通常用于链接预测)。
现在,我们知道了如何处理自同构,实现用于链接预测的单图 FM 的唯一一步就是增加对异构节点特征的支持——也许 GraphAny 风格的方法可能会带来启发?
📚更多内容请阅读参考文献 [4][5][6][7]
知识图谱推理:ULTRA 和 UltraQuery
知识图谱具有特定于图的实体和关系集,例如 Wikipedia/Wikidata 中的常见百科全书事实或 Hetionet 中的生物医学事实,这些关系具有不同的语义,并且不能直接相互映射。多年来,KG 推理模型被硬编码到给定的关系词汇表中,无法转移到具有全新实体和关系的新的、未见过的 KG。
ULTRA https://openreview.net/forum?id=jVEoydFOl9是第一个在推理时以零样本方式迁移到任何 KG 的 KG 推理基础模型。也就是说,单个预训练模型可以对具有任何大小和实体/关系词汇的任何多关系图进行推理。ULTRA 的平均成绩超过 57 个图,其表现明显优于专门针对每个图进行训练的基线。最近,ULTRA 扩展到UltraQuery https://arxiv.org/abs/2404.07198,以支持涉及连词、析取词和否定运算符的图上更复杂的逻辑查询。UltraQuery 可以迁移到看不见的图和这些看不见的图上的 10 多个复杂查询模式,其表现优于从头开始训练的更大的基线。
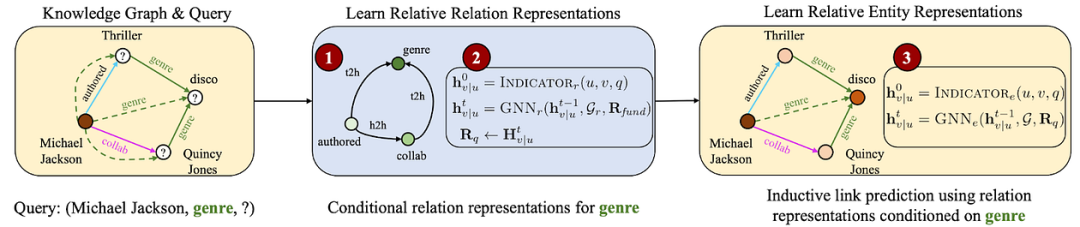
给定一个查询(迈克尔·杰克逊、流派、?),ULTRA 会构建一个关系图(边类型),以捕获原始图中以查询关系(流派)为条件的交互,并从这个较小的图中得出关系表示。然后,这些特征被用作原始较大图中的边类型特征来回答查询。
设置:给定一个具有 |E| 个节点和 |R| 个边类型的多关系图 G,没有节点特征,通过返回给定图中所有节点的概率分布来回答简单的 KG 完成查询(头部、关系、?)或涉及逻辑运算符的复杂查询。节点和关系类型的集合取决于图,并且可能会有所不同。
可迁移性:ULTRA 依赖于关系交互建模。暂时忘记关系身份和目标图域,如果我们发现“作者”和“合作”关系可以共享相同的起始节点,而另一个图中的“学生”和“合著者”关系可以共享起始节点,那么这两对关系的相对结构表示可能相似。这适用于任何领域中的任何多关系图,无论是百科全书还是生物医学 KG。ULTRA 更进一步,捕获了关系之间的 4 种此类“基本”交互。这些基本交互可以迁移到任何 KG(连同学习到的 GNN 权重)——这样,一个预先训练好的模型就可以对任何看不见的图和简单或复杂的推理查询进行推理。
📚更多内容请阅读参考文献 [8][9]
算法推理:通才算法学习者
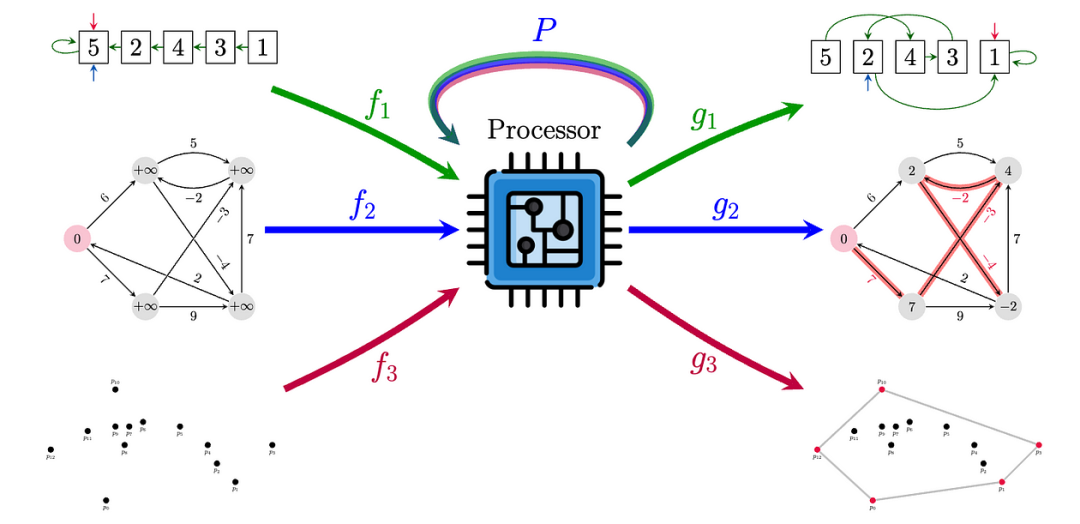
通用神经算法学习器是单处理器 GNN P,具有一组权重,能够在共享潜在空间中解决多个算法任务(每个任务都通过简单的编码器/解码器 f 和 g 连接到 P)。除此之外,处理器网络还能够进行排序(顶部)、最短路径查找(中间)和凸包查找(底部)。
设置:神经算法推理(NAR) https://arxiv.org/abs/2105.02761 研究标准算法(例如排序、搜索、动态规划)在潜在空间中的执行以及对任意大小输入的泛化。许多此类算法可以用图形输入和指针表示。给定具有节点和边特征的图 G,任务是模拟算法并产生正确的输出。或者,您可以访问提示——算法中间状态的时间序列,可作为中间监督信号。显然,不同的算法需要执行不同数量的步骤,因此这里的长度不是固定的。
可迁移性:相似算法的同质特征空间和相似控制流。例如,Prim 算法和 Dijkstra 算法具有相似的结构,仅在关键函数和边缘松弛子程序的选择上有所不同。此外,还有几个证据https://arxiv.org/abs/2203.15544表明消息传递和动态规划之间存在直接关联。这是“处理器”神经网络的主要动机,该网络可更新所有考虑算法的潜在状态(CLRS 书中的30 种经典算法https://github.com/google-deepmind/clrs)。
Triplet-GMPNN https://proceedings.mlr.press/v198/ibarz22a/ibarz22a.pdf是第一个这样的通用处理器神经网络(到 2024 年,它在 NAR 文献中已成为相当标准的神经网络)——它是一个对节点三元组及其特征进行操作的 GNN(类似于AlphaFold 中的Edge Transformers https://arxiv.org/abs/2112.00578和三角注意力)。该模型在基准测试中的所有算法任务上以多任务模式进行训练,并采用一些优化和技巧。与单任务专家模型相比,单个模型将 30 个任务的平均性能提高了 20% 以上(绝对数字)。
尽管如此,编码器和解码器都是针对每个任务专门参数化的——统一输入和输出格式的方法之一可能是使用 LLM 处理器的文本,就像在最近的CLRS 文本版本https://arxiv.org/abs/2406.04229中所做的那样。
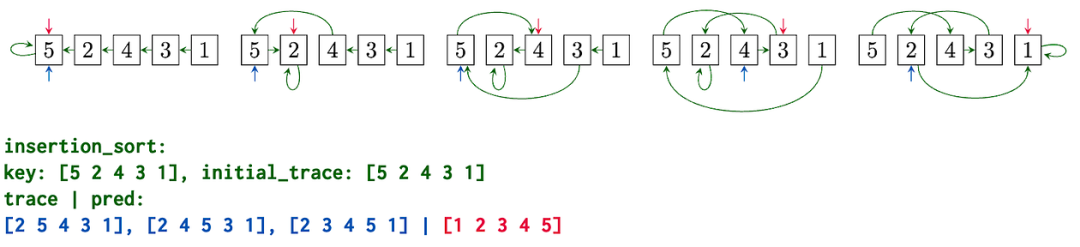
顶部:以图形形式呈现的对列表[5, 2, 4, 3, 1]进行插入排序的算法轨迹。底部:使用 CLRS-Text 生成器以文本形式呈现的相同算法轨迹。模型接收输入数组 (key) 和排序轨迹 (initial_trace) 的初始值作为输入 (以绿色表示),使用该输入提示模型预测逐步对列表进行排序的轨迹 (以蓝色表示),即从左到右将一个元素一次插入到部分排序的列表中。最后,模型需要输出最终排序后的数组 (以红色表示),并评估该数组是否预测正确。
也许 2024 年和 2025 年 NAR 最有趣的问题是:
OOD 泛化的算法推理思想能否成为可泛化的 LLM 推理的关键?
众所周知,法学硕士在处理复杂推理问题时会遇到困难,每月有数十篇论文出现在 arxiv 上,尝试一种新的提示方法,将基准测试性能提高一到两个百分点,但大多数论文无法在具有类似图结构的任务之间迁移(见下面的示例)。我们需要更多原则性的方法,而 NAR 有可能填补这一空白!

LLMs在具有类似图结构的推理问题上失败。
📚更多内容请阅读参考文献 [10][11]
几何和 AI4Science 基础模型
在几何深度学习和科学应用领域,基础模型正逐渐成为通用 ML 潜力、蛋白质语言模型和通用分子特性预测器。尽管在大多数情况下都存在通用词汇(例如小分子中的原子类型或蛋白质中的氨基酸),而且我们不必考虑通用特征化,但主要的复杂性在于原子对象在现实世界中的物理性质——它们具有明显的 3D 结构和特性(如能量),这些理论依据植根于化学、物理学和量子力学。
ML 势:用于分子的 JMP-1、DPA-2、用于无机晶体的 MACE-MP-0 和 MatterSim
设置:给定一个 3D 结构,预测结构的能量和每个原子的力;
什么是可转移的:来自元素周期表的原子词汇。
ML 势能根据化合物(如分子或周期性晶体)的 3D 坐标和可选输入(如晶体的周期性边界条件)估算化合物的势能。对于任何原子模型,可能的原子词汇始终受元素周期表的约束,元素周期表目前包含 118 种元素。ML 势能的“基础”方面是推广到任何原子结构(可以有多种组合),并且足够稳定,可用于分子动力学 (MD)、药物和材料发现流程。
大约在同一时间发布的JMP-1 https://arxiv.org/abs/2310.16802和DPA-2 https://arxiv.org/abs/2312.15492旨在成为这种通用的 ML 潜在模型——它们在各种各样的结构上进行训练——从有机分子到晶体再到 MD 轨迹。例如,单个预训练的 JMP-1 在 QM9、小分子的 rMD17、晶体的 MatBench 和 QMOF 以及大分子的 MD22、SPICE 方面表现出色,与专门的数据集模型相当或更好。同样,MACE-MP-0 https://arxiv.org/abs/2401.00096和MatterSim https://arxiv.org/abs/2405.04967是最先进的无机晶体 FM(MACE-MP-0 已经提供权重),在 20 多个晶体任务上进行了评估,从多组分合金到燃烧和熔盐。等变 GNN 是这些系统的核心,有助于处理等变特征(笛卡尔坐标)和不变特征(如原子类型)。
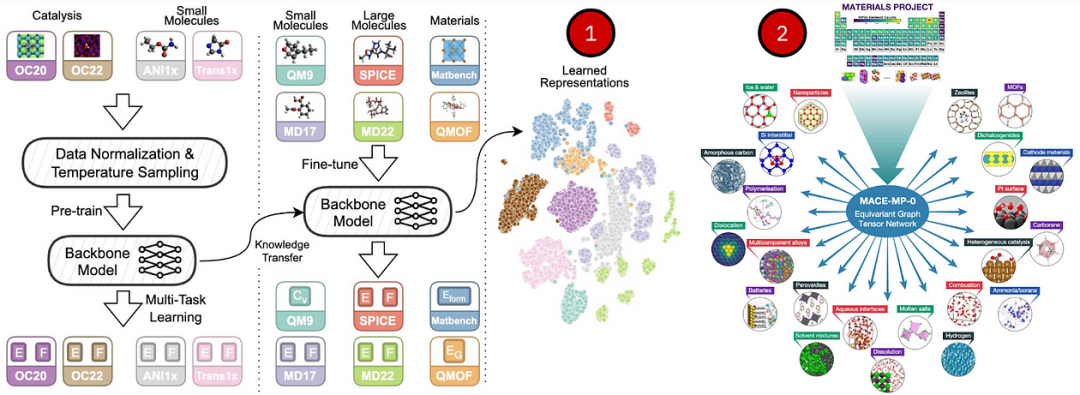
(1) JMP-1对分子和晶体的预训练和微调;(2)MACE-MP-0仅在材料项目数据上进行训练,并转移到固相、液相和气相各种化学反应的分子动力学模拟。
下一个前沿似乎是机器学习加速的分子动力学模拟——传统计算方法在飞秒级(10-15)内工作,需要数百万和数十亿步来模拟分子、晶体或蛋白质。加快此类计算将产生巨大的科学影响。
📚更多内容请阅读参考文献 [12][13][14][15]
蛋白质 LM:ESM-2
设置:给定一个蛋白质序列,预测类似于掩蔽语言建模的掩蔽标记;
可转移的词汇量:20(22)种氨基酸。
蛋白质序列类似于以氨基酸作为标记的自然语言,而 Transformers 擅长编码序列数据。尽管氨基酸的词汇量相对较小,但可能的蛋白质空间却非常巨大,因此对大量已知蛋白质进行训练可能会暗示未见过的组合的特性。由于预训练数据量大、各种可用检查点和信息丰富的特征, ESM-2 https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1可能是最受欢迎的蛋白质 LM 。
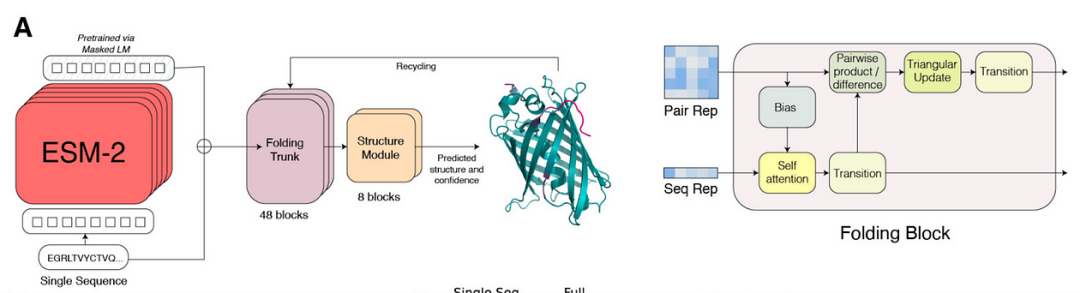
ESM2 作为掩蔽 LM 和 ESMFold 用于蛋白质结构预测。
ESM 特征用于无数应用,从预测 3D 结构(在ESMFold https://github.com/facebookresearch/esm中)到蛋白质-配体结合(DiffDock https://arxiv.org/abs/2210.01776及其后代)到蛋白质结构生成模型(如最近的FoldFlow 2 https://www.dreamfold.ai/blog/foldflow-2)。更大的 Transformer 和更多的数据可能会进一步提高蛋白质 LM 的性能——然而,在这个规模下,数据问题变得更加普遍(我们还在专门部分讨论了架构和数据之间的相互作用),例如,ESM Metagenomic Atlas https://esmatlas.com/已经编码了 7 亿多个结构,包括在土壤、海洋或热液喷口中人类外部所见的结构。有没有办法像常见的 LLM 训练数据集那样获得数万亿个 token?
📚更多内容请阅读参考文献 [16][17]
2D 分子:MiniMol 和 MolGPS
设置:给定具有原子类型和键类型的二维图形结构,预测分子特性。
什么是可转移的:元素周期表中的原子和键类型的词汇。
对于 2D 图(没有 3D 原子坐标),通用编码和可转移性来自固定的原子和键类型词汇表,您可以将其发送到任何 GNN 或 Transformer 编码器。尽管自 1960 年代以来就已使用分子指纹(Morgan fingerprints摩根指纹[18] https://pubs.acs.org/doi/abs/10.1021/c160017a018),但它们的主要目标是评估相似性,而不是对潜在空间进行建模。单个(大型)神经编码器的任务是学习可能暗示某些物理分子特性的有用表示。
用于学习分子表征的通用模型的最新示例是MiniMol https://arxiv.org/pdf/2404.14986和MolGPS https://arxiv.org/abs/2404.11568v1,它们已在大量分子图语料库上进行训练,并在数十个下游任务上进行了探索。也就是说,您仍然需要根据模型的表征微调单独的特定于任务的解码器/预测器——从这个意义上说,一个预先训练的模型将无法对所有可能的未见任务运行零样本推理,而是对解码器已训练过的任务运行零样本推理。不过,微调仍然是一个很好的廉价选择,因为这些模型比 LLM 小几个数量级。
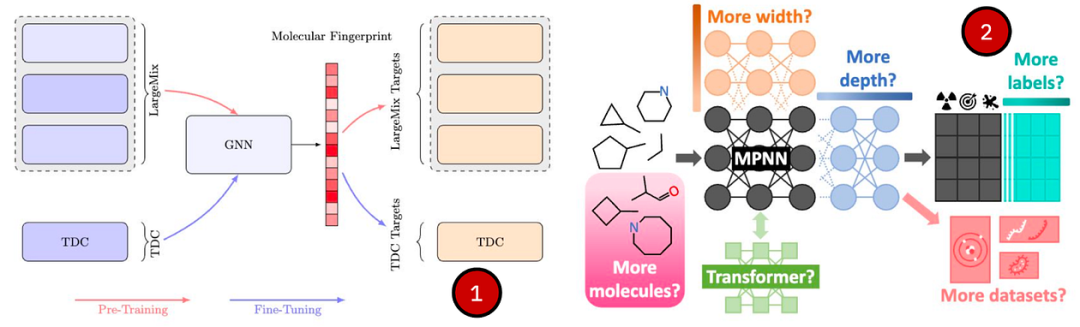
(1)MiniMol预训练和下游任务评估的工作流程概述。
(2)MolGPS扩展研究的标准
📚更多内容请阅读参考文献 [19][20]
表达力与缩放定律:图 FM 是否可以缩放?
LLM 和多模态边界模型中的 Transformer 相当标准,我们知道它们的一些基本扩展原则。Transformer(作为一种架构,而不是 LLM)在图上是否同样有效?在为 Graph FMs 设计骨干时,一般面临的挑战是什么?
如果对上文中重点介绍的模型进行分类,只有两个领域具有 transformers——具有自然序列偏差的蛋白质 LMs(ESM)和小分子(MolGPS)。其余的是 GNN。原因如下:
-
普通 Transformer 无法扩展到任何大于标准上下文长度(>4-10k 个节点)的合理大图。任何超过该范围的情况都需要一些技巧,例如仅提供子图(丢失整个图结构和长距离依赖关系)或线性注意(可能不具有良好的扩展性)。相比之下,GNN 在边数上是线性的,在稀疏图(V ~ E)的情况下,在节点数上是线性的。
-
没有位置编码的 Vanilla Transformer 的表达能力不如 GNN https://arxiv.org/abs/2302.04181。在具有 V 节点的图上挖掘像拉普拉斯 PE 这样的位置编码是 O(V³)。
-
通过 transformer 编码图时,“token” 应该是什么?文献中没有明确的赢家,例如,节点https://arxiv.org/abs/2106.05234、节点 + 边https://arxiv.org/abs/2406.03148或子图https://arxiv.org/abs/2212.13350都是可行的选择。
➡️谈到表达力,不同的图任务需要处理不同的对称性,例如,链接预测中的自同构节点导致难以区分的表示,而在图分类 / 回归中,超越 1-WL 是区分分子所必需的,否则这些分子可能看起来与原始 GNN 同构。
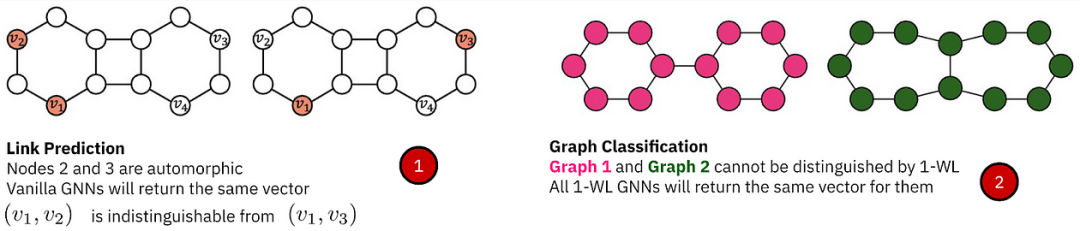
不同的任务需要处理不同的对称性。
这一事实引出了两个问题:
GFM 应该有多强的表现力?表现力和可扩展性之间的权衡是什么?
理想情况下,我们希望单个模型能够同样出色地解决所有这些对称性问题。然而,更具表现力的模型会导致在训练和推理方面架构的计算成本更高。我们同意最近 ICML'24关于 Graph ML 理论未来方向的立场文件https://arxiv.org/abs/2402.02287,即社区应该在表现力、泛化和优化之间寻求平衡。
不过,值得注意的是,随着训练数据的可用性不断提高,推迟直接从数据中学习复杂的对称性和不变性(而不是将它们烘焙到模型中)可能是一种计算成本更低的想法。最近几个很好的例子是AlphaFold 3 https://www.nature.com/articles/s41586-024-07487-w和Molecular Conformer Fields https://arxiv.org/abs/2311.17932,它们在许多生成应用中达到了 SOTA,而无需昂贵的等变几何编码器。
📚更多内容请阅读参考文献 [21]
➡️当谈到扩展时,模型和数据都应该扩大。然而:
❌ 非几何图:目前还没有关于将 GNN 或 Transformer 扩展到大图和常见任务(如节点分类和链接预测)的原则性研究。2 层 GraphSAGE 通常与巨大的 16 层图 Transformer 相差不远。类似的趋势是,在 KG 推理领域,一个参数少于 200k 的 ULTRA 模型(如上所述)在 50 多个图上的表现优于数百万大小的浅嵌入模型。为什么会这样?我们假设关键在于 1️⃣ 任务性质——大多数非几何图都是噪声相似图,不受分子等具体物理现象的约束 2️⃣ 给定丰富的节点和边特征,模型必须学习图结构的表示(常用于链接预测)或仅仅是给定特征的函数(一个很好的例子是OGB 中的节点分类https://ogb.stanford.edu/docs/leader_nodeprop/,其中大多数收益是通过添加 LLM 特征编码器来实现的)。
✅ 几何图:最近有几篇研究重点关注分子图:
-
Frey 等人(2023)研究了几何 GNN 对 ML 潜力的扩展;
-
Sypetkowski、Wenkel 等人(2024 年)引入 MolGPS,并研究在 500 万个分子的大型数据集上将 MPNN 和 Graph Transformers 扩展到 10 亿个参数;
-
Liu 等人(2024) 在多达 400 万个分子的分子数据集上探测了 GCN、GIN 和 GraphGPS 多达 1 亿个参数。
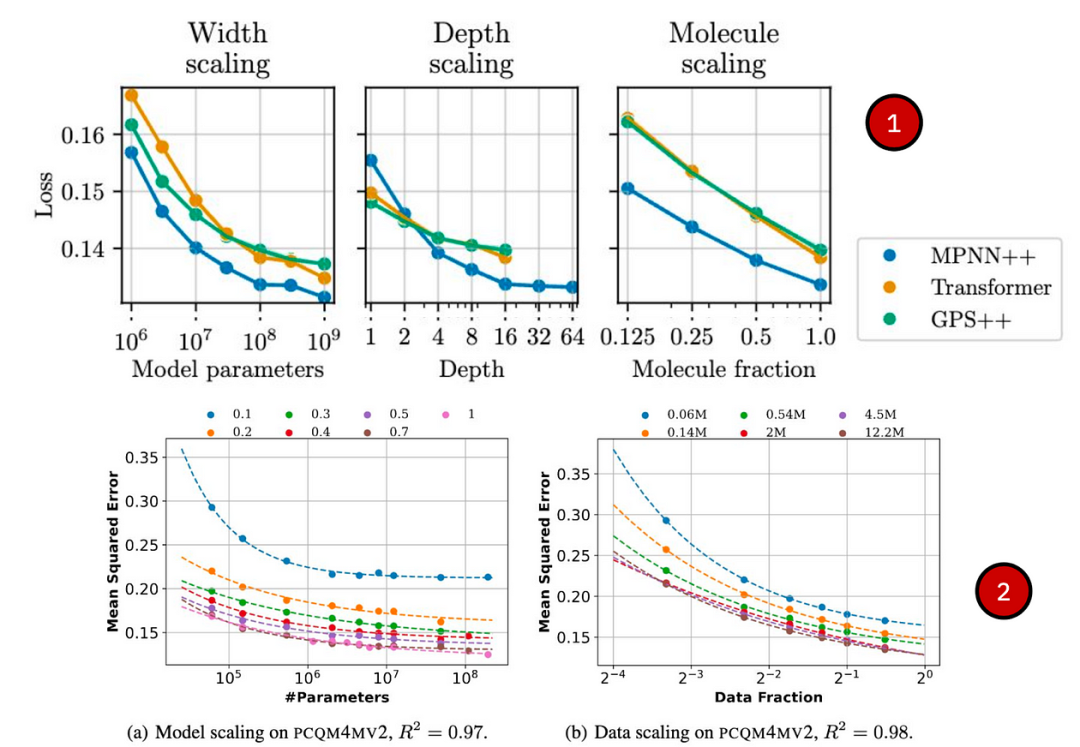
缩放分子 GNN 和 GT。
数据问题:应该缩放什么?是否有足够的图形数据来训练 Graph FMs?
1️⃣图形数据中应该缩放什么?节点?边?图形的数量?还有别的吗?
文献中没有明确的赢家,我们更倾向于使用更广泛的术语多样性,即图数据中的模式多样性。例如,在大型产品图上的节点分类中,如果你在具有 1 亿个节点或 100 亿个节点的图上进行训练,可能不会有太大影响,因为它与用户项目图的性质相同。但是,展示不同规模和稀疏度的同质性和异质性的示例可能会非常有益。在GraphAny中,展示此类图的示例可以构建一个可推广到不同图分布的稳健节点分类器,
在使用ULTRA进行 KG 推理时,发现预训练中关系模式的多样性对归纳泛化的影响最大,例如,一个大的密集图比一组较小但稀疏的、密集的、少关系的和多关系的图更差。
在分子图级任务中,例如在MolGPS中,扩展具有不同物理特性的独特分子的数量有很大帮助(如上图所示)。
此外,UniAug https://arxiv.org/abs/2406.01899发现,预训练数据中结构模式的覆盖率越高,越能提高各个领域的不同下游任务的性能。
2️⃣ 有足够的数据来训练 Graph FMs 吗?
公开可用的图形数据比自然语言标记、图像或视频要小几个数量级,而且没问题。本文包含数千个语言和图像标记,但没有明确的图形(除非您尝试将此文本解析为类似于抽象含义表示图https://en.wikipedia.org/wiki/Abstract_Meaning_Representation的图)。PDB 中具有已知结构的“好”蛋白质数量很少,已知的“好”药物分子数量也很少。
Graph FMs 是否会因为数据稀缺而注定失败?
其实并非如此。两个可行的途径是:(1)更高效的采样架构;(2)使用更多黑盒和合成数据。
GraphWorld https://arxiv.org/abs/2203.00112等合成基准可能有助于增加训练数据的多样性,并提高对真实世界数据集的泛化能力。反过来,从科学实验中获得的黑盒数据很可能成为在 AI 4 Science 中构建成功基础模型的关键因素——掌握它的人将在市场上占据优势。
📚更多内容请阅读参考文献 [20][22][23]
关键要点
➡️如何跨具有异构节点/边/图特征的图进行概括?
-
非几何图:相对信息传递(例如GraphAny中的预测差异或Ultra中的关系交互),绝对信息则不传递。
-
几何图:由于原子集固定,转移变得更容易,但模型必须学习一些物理概念才能可靠。
➡️如何在预测任务中进行概括?
-
迄今为止,还没有单一模型(在非几何 GNN 中)能够在零样本推理模式下执行节点分类、链接预测和图分类。
-
通过一个视角来构建所有任务可能会有所帮助,例如,节点分类可以被构建为链接预测。
➡️ 最优模型表达力是多少?
-
节点分类、链接预测和图分类利用不同的对称性。
-
最大限度表达模型的粗略应用会很快导致指数级的运行时复杂性或巨大的内存成本——需要保持expressivity表达力与efficiency效率的平衡。
-
表达力、样本复杂性(需要多少训练数据)和归纳泛化之间的联系仍然未知。
➡️ 数据
-
公开可用的图形数据比文本/视觉数据小几个数量级,模型必须具有样本效率。
-
Scaling laws缩放定律正处于新兴阶段,缩放什么仍不清楚——nodes节点数?Edges边?Motifs图案?图中的标记概念是什么?
-
几何 GNN:有更多的实验数据可用,这些数据对于领域专家来说意义不大,但可能对神经网络有价值。
参考
-
Mao, Chen, et al. Graph Foundation Models Are Already Here. ICML 2024
-
Morris et al. Future Directions in Foundations of Graph Machine Learning. ICML 2024
-
Zhao et al. GraphAny: A Foundation Model for Node Classification on Any Graph. Arxiv 2024. Code on Github
-
Dong et al. Universal Link Predictor By In-Context Learning on Graphs, arxiv 2024
-
Zhang et al. Labeling Trick: A Theory of Using Graph Neural Networks for Multi-Node Representation Learning. NeurIPS 2021
-
Chamberlain, Shirobokov, et al. Graph Neural Networks for Link Prediction with Subgraph Sketching. ICLR 2023
-
Zhu et al. Neural Bellman-Ford Networks: A General Graph Neural Network Framework for Link Prediction. NeurIPS 2021
-
Galkin et al. Towards Foundation Models for Knowledge Graph Reasoning. ICLR 2024
-
Galkin et al. Zero-shot Logical Query Reasoning on any Knowledge Graph. arxiv 2024. Code on Github
-
Ibarz et al. A Generalist Neural Algorithmic Learner LoG 2022
-
Markeeva, McLeish, Ibarz, et al. The CLRS-Text Algorithmic Reasoning Language Benchmark. arxiv 2024
-
Shoghi et al. From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction. ICLR 2024
-
Zhang, Liu et al. DPA-2: Towards a universal large atomic model for molecular and material simulation, arxiv 2023
-
Batatia et al. A foundation model for atomistic materials chemistry, arxiv 2024
-
Yang et al. MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures, arxiv 2024
-
Rives et al. Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences. PNAS 2021
-
Lin, Akin, Rao, Hie, et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. Science 2023. Code
-
Morgan HL (1965) The generation of a unique machine description for chemical structures — a technique developed at chemical abstracts service. J Chem Doc 5:107–113
-
Kläser, Banaszewski, et al. MiniMol: A Parameter Efficient Foundation Model for Molecular Learning, arxiv 2024
-
Sypetkowski, Wenkel et al. On the Scalability of GNNs for Molecular Graphs, arxiv 2024
-
Morris et al. Future Directions in Foundations of Graph Machine Learning. ICML 2024
-
Liu et al. Neural Scaling Laws on Graphs, arxiv 2024
-
Frey et al. Neural scaling of deep chemical models, Nature Machine Intelligence 2023
编辑:黄继彦
关于我们
数据派THU作为数据科学类公众号,背靠清华大学大数据研究中心,分享前沿数据科学与大数据技术创新研究动态、持续传播数据科学知识,努力建设数据人才聚集平台、打造中国大数据最强集团军。

新浪微博:@数据派THU
微信视频号:数据派THU
今日头条:数据派THU

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐
 0
0 0
0- 0



 已为社区贡献169条内容
已为社区贡献169条内容

所有评论(0)