“深入解析蛋白-配体模拟:从对接到动力学模拟与自由能计算的完整流程” Gromacs 蛋白-配体体系模拟 MD思路整理 思维导图 Gromacs MD出结果后可得到那些结果
引言:
之前讲过了 Gromacs膜蛋白的模拟流程
[GROMACS 蛋白-配体分子动力学模拟]
在里面描述了进行MD模拟 核心命令 思路顺序,接到一些读者的疑惑,下面重新整理一下Gromacs 进行MD的思维顺序——附赠思维导图。这篇博客将详细介绍如何通过集成不同的软件工具,逐步完成一个完整的蛋白-配体体系模拟流程。
一、涉及的核心软件
- Gromacs2025.3 GPU加速额外集成 MPI 并行加速版
经过配置显卡策略,RTX5070Ti 显卡 跑200ns 用时 5 hours 10 minutes
- AmberTools25 GPU加速额外集成 MPI 并行加速版
- Amber24(Pmemd24) GPU加速额外集成 MPI 并行加速版
- OpenBabel
- MGLTOOLS
- Vina-GPU 10层逆向叠加编译
- Chem3d (ChemOffice三件套)
- gmxMMPBSA
- Grace 可用可不用(精通Grace者可优先选择)
二、蛋白-配体体系模拟思路
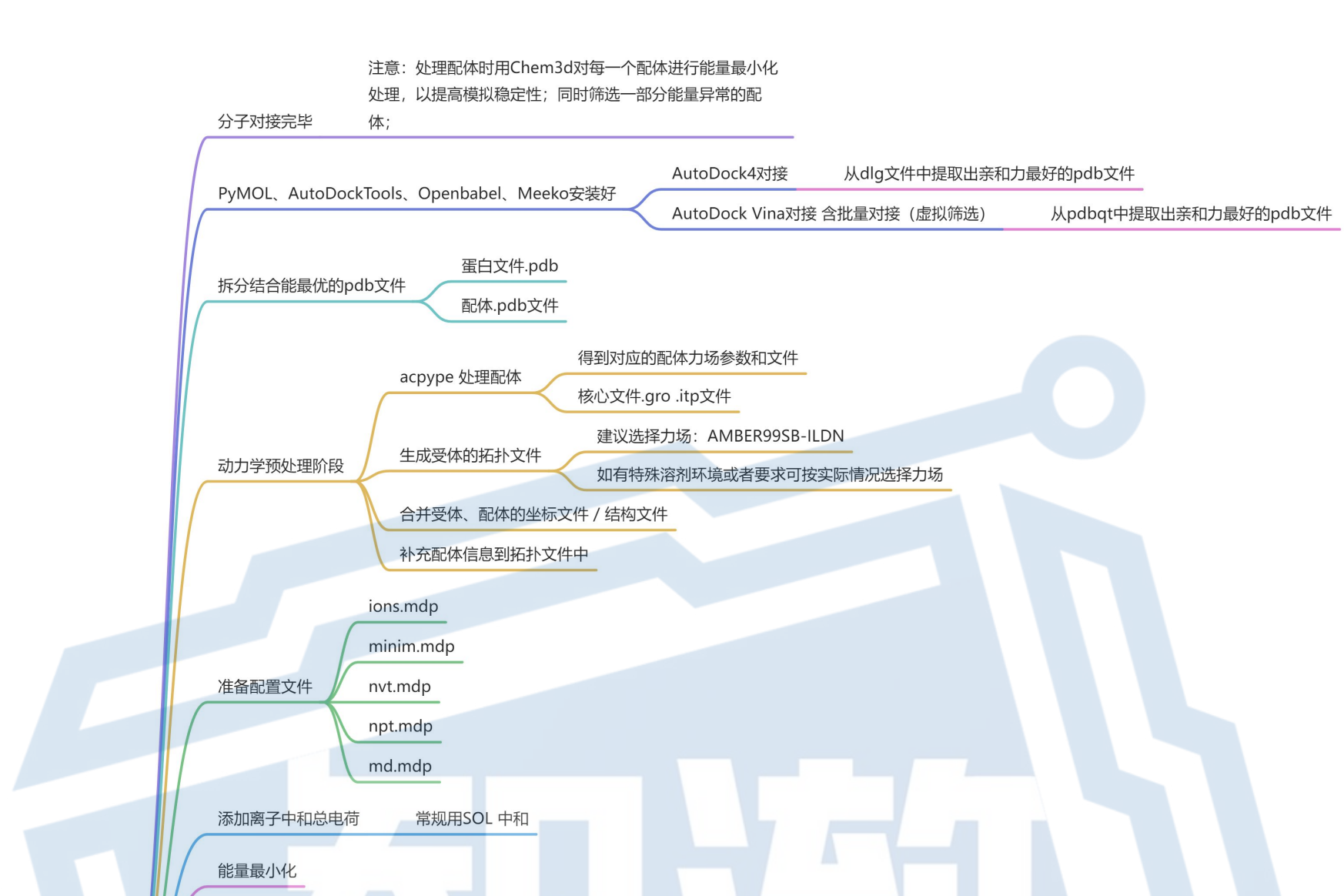
1. 分子对接完毕
注意:处理配体时用Chem3d对每一个配体进行能量最小化处理,以提高模拟稳定性;同时筛选一部分能量异常的配体;
2. 安装基础软件
PyMOL、AutoDockTools、Openbabel、Meeko安装好
注意:AutoDock4对接和AutoDock Vina对接 含批量对接(虚拟筛选)是两种方式,结果文件是不一样的
3. 提取出亲和力最好的结果文件

4. 拆分结合能最优的pdb文件
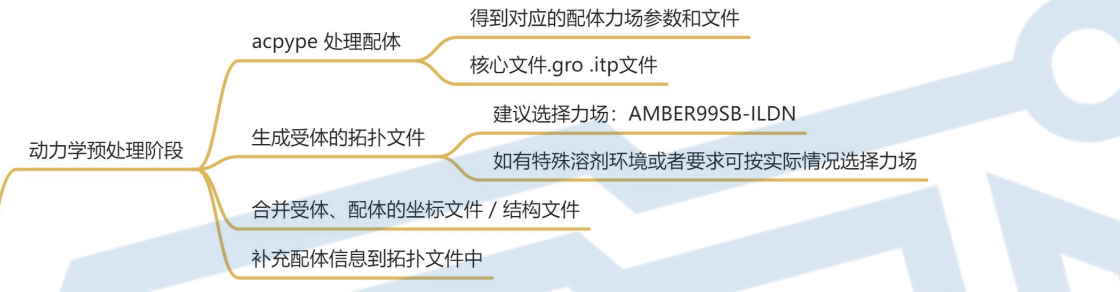
5. 动力学预处理阶段
-
acpype 处理配体
-
生成受体的拓扑文件
-
合并受体、配体的坐标文件 / 结构文件
-
补充配体信息到拓扑文件中
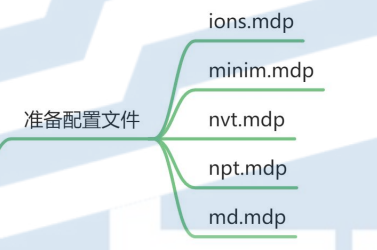
6. 准备配置文件
7. 添加离子中和总电荷
系统中通常需要添加离子以中和总电荷,确保系统的电荷平衡,这对于后续的能量最小化和动力学模拟至关重要。
8. 能量最小化
在进行分子动力学模拟之前,能量最小化是必不可少的步骤。它帮助消除系统中的不合理高能构象,避免模拟过程中出现大幅度的非物理行为。
9. 系统平衡
-
NVT平衡
-
NPT 平衡
10. 进行动力学生产模拟 MD
平衡完成后,可以开始进行生产模拟。在这一阶段,分子系统将在目标条件下进行长时间的动力学模拟,通常持续数百纳秒,以确保足够的采样。
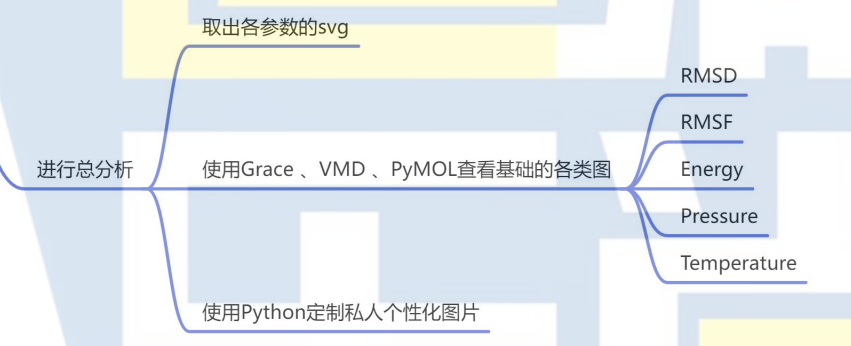
11. 进行总分析
12. 按情况对有无氢键进行处理
-
有氢键:对氢键进行分析,了解氢键对蛋白质-配体结合的贡献。
-
无氢键连接:如果没有氢键作用,需要特别关注疏水作用力和其他非共价相互作用。

13. 算 MM/PBSA + 残基分解
gmxMMPBSA:单线程
MMPBSA.MPI:可配置MPI并行计算
14. PCA分析
基于分子的运动和主成分分析(PCA),得到2D、3D自由能景观图;
15. 提供pdf思维导图原图
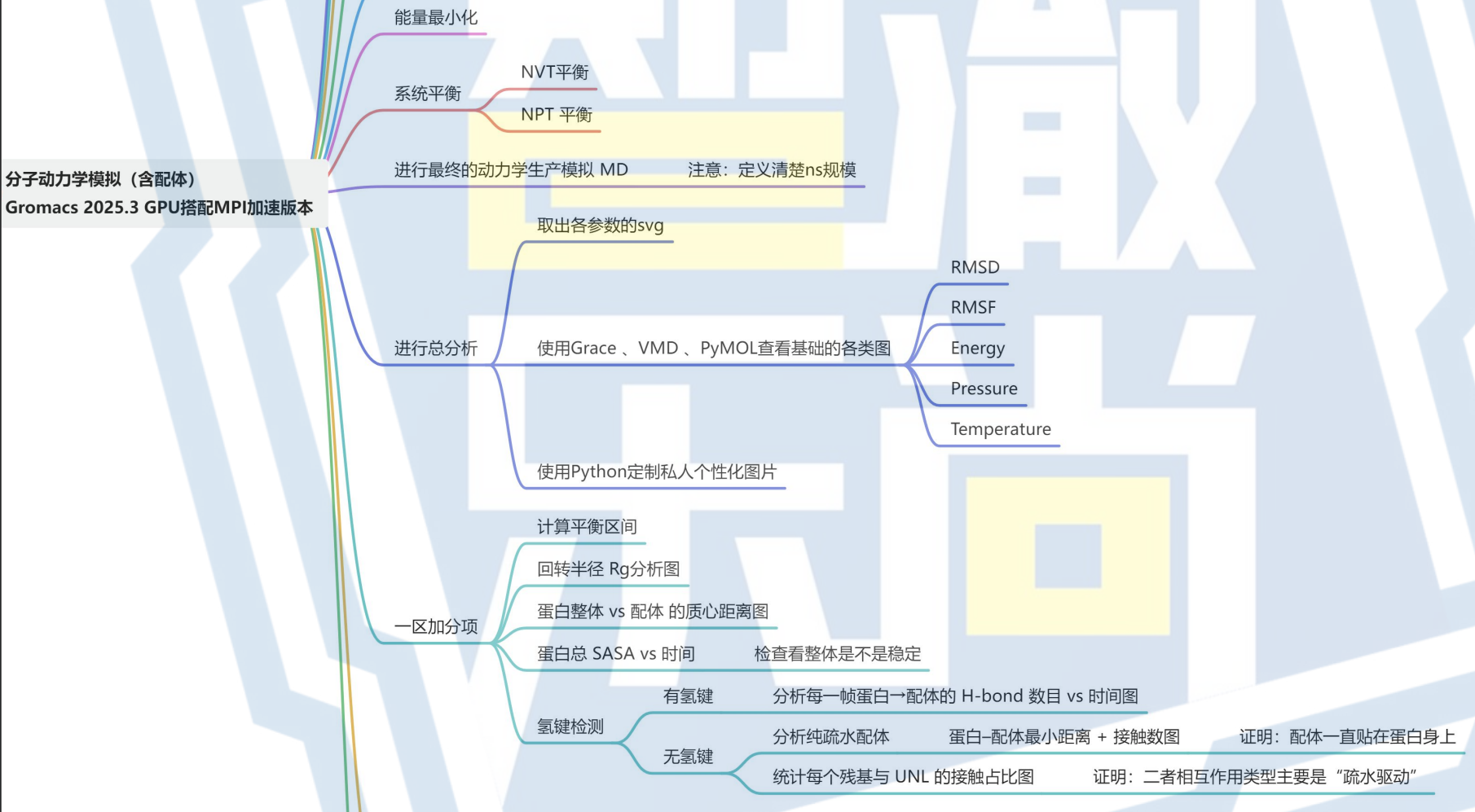
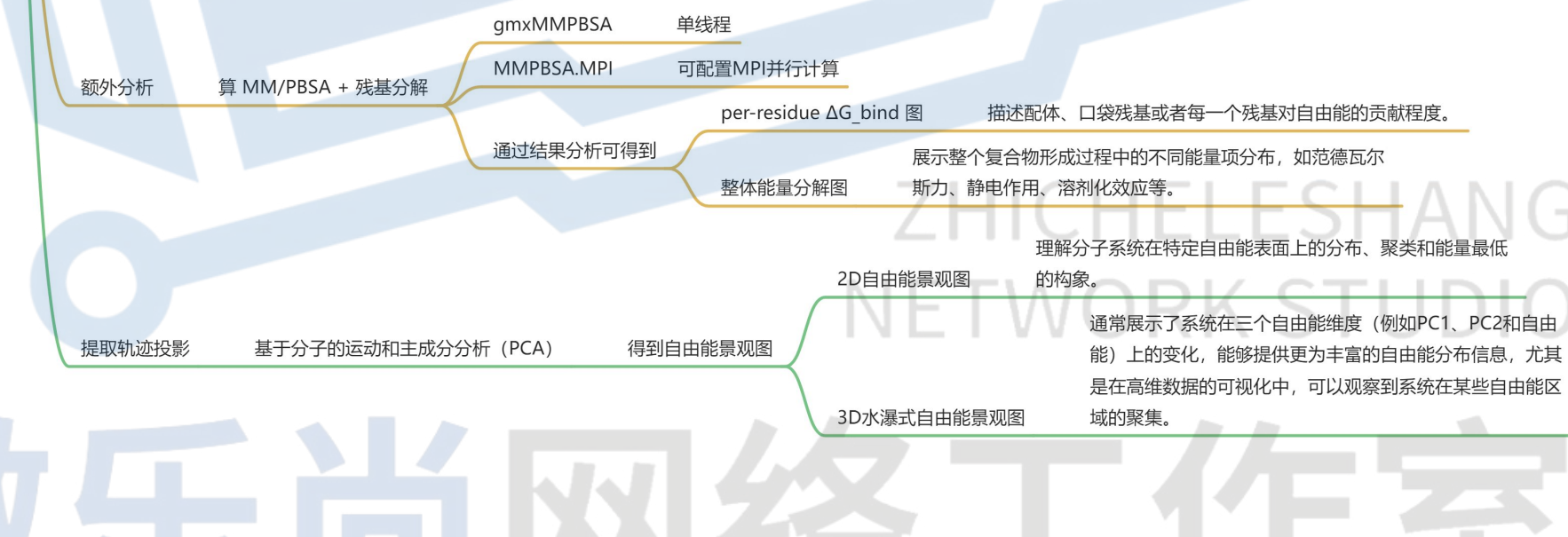
有需要完整的pdf文件的友友,可以直接联系我
这里不太好整个完整截图!!!
三、结果分析与验证
在进行分子动力学模拟和自由能计算之后,结果的分析和验证是确保模拟准确性和可靠性的关键步骤。以下是如何分析和验证模拟结果的一些关键方面:
1. 结合能分析
结合能是蛋白-配体结合的重要指标。通过计算每个构象的结合能,可以评估配体与蛋白质的亲和力。常见的结合能分析方法包括MM/PBSA(分子力场/泊松-玻尔兹曼表面区分法)和MM/GBSA(分子力场/广义玻尔兹曼表面区分法)。这些方法可以帮助进一步确认最优结合模式并排除不合理的构象。
2. 氢键网络分析
如果模拟体系中存在氢键作用,需要详细分析氢键的数量、位置和稳定性。氢键是蛋白-配体结合的常见相互作用之一,其强度和稳定性在分子动力学模拟过程中可能发生变化。分析氢键的形成和断裂规律有助于理解配体的结合机制。
3. 动力学特征分析
-
RMSD(均方根偏差):通过计算蛋白和配体的RMSD,可以监测系统在模拟过程中是否发生了显著的结构变化。如果RMSD保持稳定,说明体系已经达到平衡状态。
-
RMSF(均方根波动):RMSF值反映了蛋白质或配体中各个残基的动态变化。较大的RMSF值通常表明该区域的结构较为灵活,而较小的值则表明该区域相对刚性。
-
氢键保持时间:分析氢键的生命周期可以帮助理解配体与蛋白质的结合稳定性。通过分析每个氢键的持续时间,可以更好地评估氢键在配体结合过程中的重要性。
4. 疏水作用力分析
疏水作用力是配体和蛋白质结合的一个重要驱动力,尤其是在没有明显氢键的情况下。在此部分,您可以利用软件工具(如PLIP或PyMOL)分析配体和蛋白之间的疏水相互作用。通过这些分析,可以了解哪些氨基酸残基主导了疏水相互作用,并评估其对结合的贡献。
5. 残基分解分析
MM/PBSA和MM/GBSA方法可以进一步提供每个残基在配体结合中的贡献。通过残基分解分析,您可以识别出对结合能有显著影响的关键残基。这个信息可以帮助优化配体和蛋白质之间的相互作用,进而为药物设计提供有价值的参考。
6. PCA分析与构象变化
主成分分析(PCA)有助于揭示蛋白质在配体结合过程中的大尺度构象变化。PCA可以提取模拟过程中最显著的运动模式,并将其与蛋白质的功能状态进行比较。对于多分子结合,PCA有助于评估配体如何引发蛋白质结构的变动。
7. 结果的验证与对比
为了确保模拟结果的可靠性,可以通过以下几种方式进行验证:
-
实验数据对比:与已有的实验结果(如X射线晶体学数据或NMR数据)进行对比,确认模拟的合理性。
-
文献验证:通过查阅相关文献,了解类似体系的模拟结果或实验结果,从而评估模拟结果的科学性。
通过这些分析步骤,可以确保模拟结果不仅在理论上合理,而且与实验数据一致,为后续的药物设计提供坚实的基础。
四、下一期提供 MD模拟结果的十六个核心结果图
敬请期待.....
《Gromacs从MD模拟到可视化:展示16个关键图形以揭示分子动力学的潜力 (适用于验证、论证 分子对接的配体没有氢键) 分子对接分数挺好,怎么没看到氢键连接?》
五、与我联系——享受一键教学 十六图 “思路”
了解清楚了安装之前的这些知识以后,如果嫌弃麻烦需要远程安装的友友可以联系!
PC端电脑通过
点击PC端分子对接软件合集——“能看到某宝对应的分子对接软件商品!!!。
手机淘宝通过:
点击手淘分子对接软件合集 “——能看到某宝对应的分子对接软件商品!!!

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐



 18
18 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)