1.GEO数据处理、可视化、富集
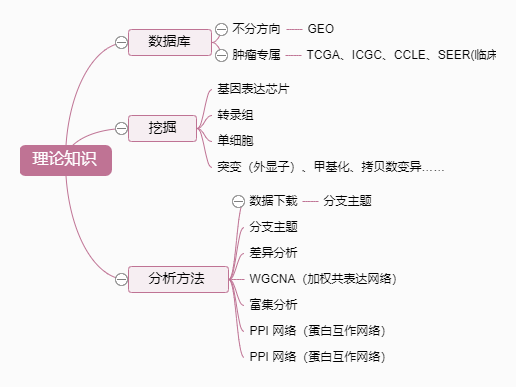
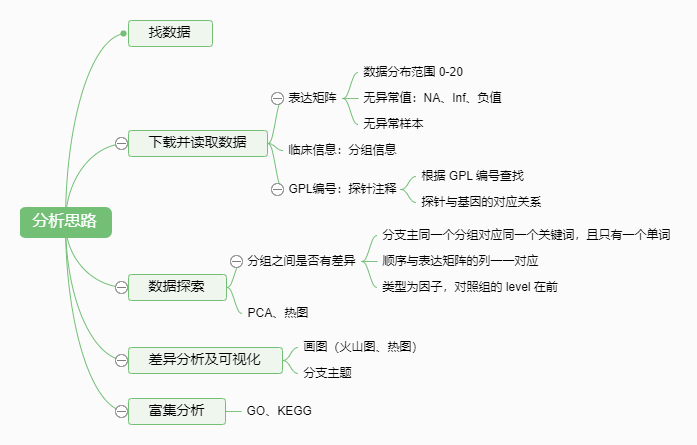
一、数据下载与读取
(GEO数据库)
1.下载
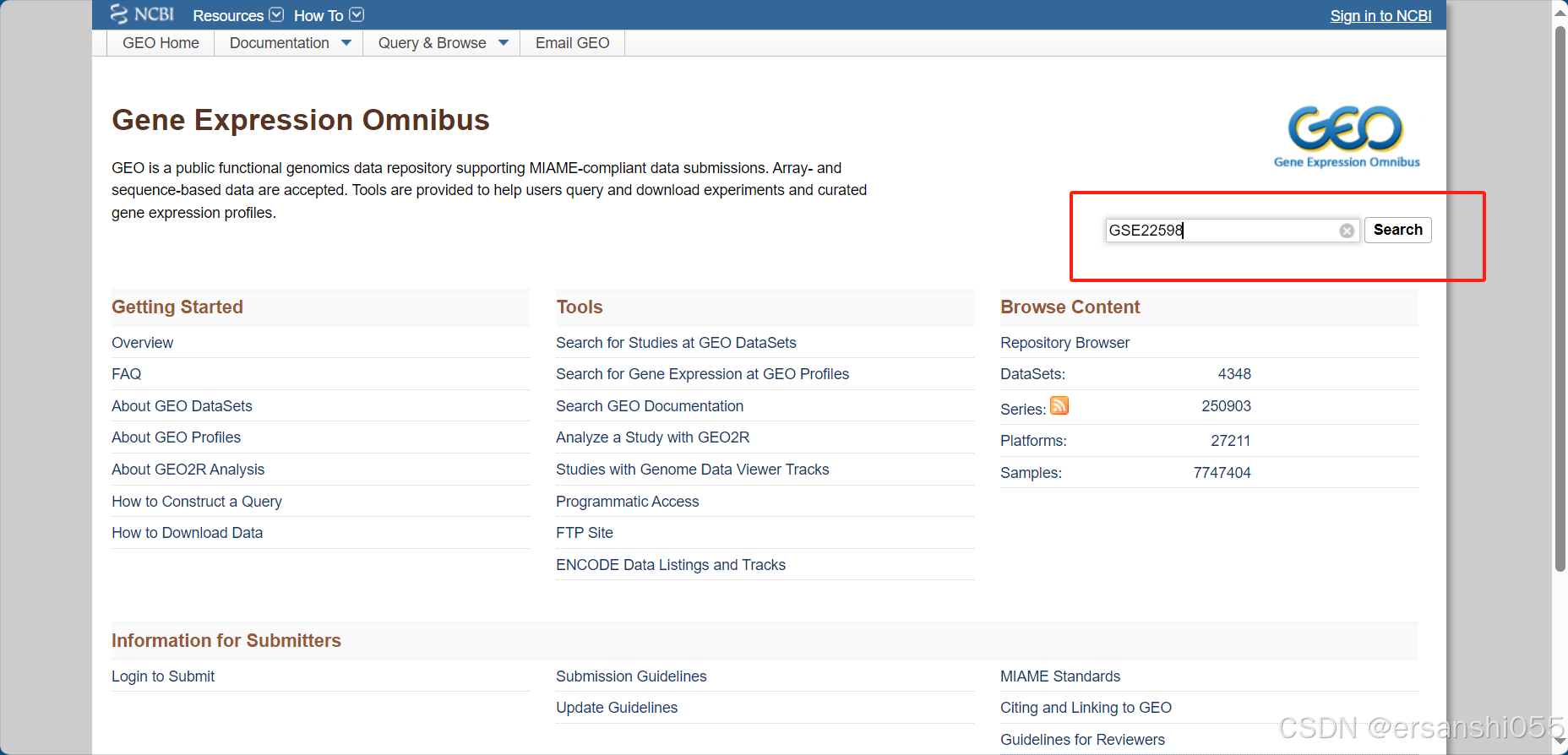
下载数据:以GSE22598为例
打开:https://www.ncbi.nlm.nih.gov/geo/,输入accession,点击search。
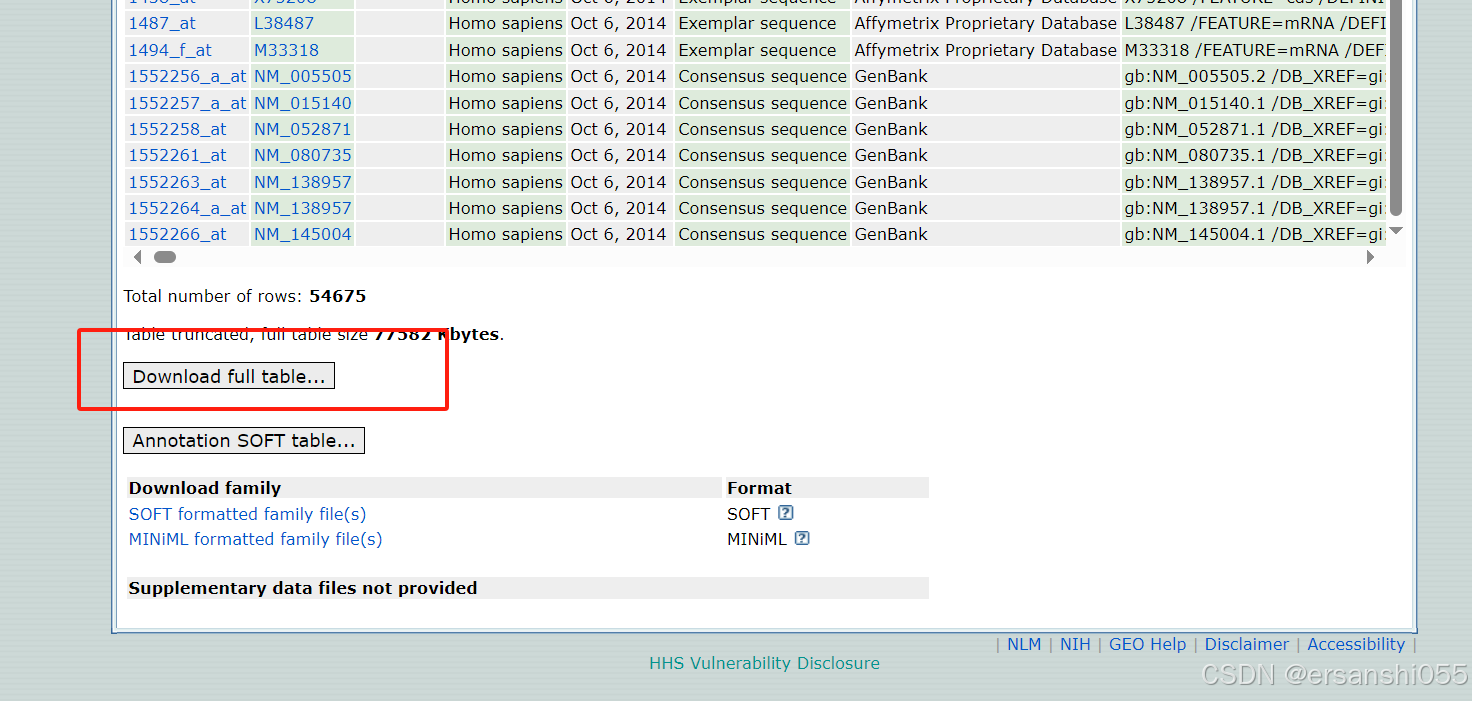
或者用代码
####---GSE22598_数据获取和读取----####
library(GEOquery) #没有下载"GEOquery"的先下载
gset <- getGEO('GSE22598', destdir = ".", AnnotGPL = F, getGPL = F) #data_download
a <- gset[[1]]
class(a)
#exprSet <- exprs(a) #获取表达数据
#Clinical <- pData(a) #获取临床信息
2.读取
有多种数据读取方法,通用的是 :
## 读取表达矩阵(最推荐的方法!!)
exprSet <- read_tsv("GSE22598_series_matrix.txt.gz",skip = 76 #跳过文件头部元数据
exprSet <- data.frame(exprSet)
class(exprSet)
## 获得行为探针ID名称,列为样本名称的表达矩阵。
rownames(exprSet) <- exprSet[,1]
exprSet <- exprSet[,-1]下载数据前,先进行以下操作,确保正常下载:
#下载数据前,先进行以下操作
####----GEO_芯片数据----####
rm(list= ls())
gc()
stringAsFactor = FALSE
getwd()
## 加载 R 包,根据理解,按照逻辑顺序逐个加载
library(readr)
# library(dplyr)
library(tidyverse)
library(ggplot2)二、芯片数据清洗
基因芯片:在一块基片表面固定序列已知的靶核苷酸探针,当溶液中带有荧光标记的核酸序列与芯片上对应位置的核酸探针产生互补配对时,通过确定荧光强度最强的探针位置,获得一组序列完全互补的探针序列。据此重组出靶核酸序列,最终得到兴趣基因或分子。
在进行探针注释前,我们需要对表达矩阵的数据进行log2转换判断和数据标准化。原理是PCR反应以2^N的方式倍增。这种处理会使得数据以更直观、简洁的方式呈现。
一般情况下,有三种方法获得探针注释信息:①金标准是从基因芯片公司官网下载。但是基因芯片平台太多,真正实操起来非常麻烦。②用bioconductor的注释包。这种方法有时要摸索很久才能获较高质量的芯片注释关系。一般情况下,芯片在bioconductor里面都是有注释包的,用户每次独立下载安装后获取。③通过GPL文件注释,类似于GSE文件的读取。主要使用tidyverse包。
1.预处理:读取数据
使用read_tsv读取的数据进行后续操作,先进行log2转换
##使用read_tsv读取的数据进行后续操作,先进行log2转换
## 1. 数据预处理:log2转换判断与执行
ex <- exprSet # 将exprSet数据赋值给ex变量,创建一个副本用于处理
# 计算关键分位数(避免重复计算)
data_quantiles <- quantile(processed_data,
probs = c(0.00, 0.25, 0.5, 0.75, 0.99, 1.0),
na.rm = TRUE)
# 定义更清晰的log2转换条件判断函数
needs_log2_transform <- function(q) {
# 条件1:数据范围大且存在高表达值
high_expression <- q[5] > 100
# 条件2:数据动态范围广且非负
wide_range <- (q[6] - q[1] > 50) && (q[2] > 0)
# 条件3:数据可能已经部分对数化
partially_logged <- (q[2] > 0 && q[2] < 1) && (q[4] > 1 && q[4] < 2)
return(high_expression || wide_range || partially_logged)
}
# 执行转换判断
if (needs_log2_transform(data_quantiles)) {
# 安全处理非正值:用NA替代而不是NaN(更符合生物数据分析惯例)
processed_data[processed_data <= 0] <- NA
# 执行log2转换并添加属性记录
processed_data <- log2(processed_data)
attr(processed_data, "transform") <- "log2"
message("Performed log2 transformation")
} else {
attr(processed_data, "transform") <- "none"
message("No transformation needed")
}
##2.数据可视化
library(limma)
# 增强的箱线图可视化函数
enhanced_boxplot <- function(data, ...) {
# 设置绘图参数
par(mar = c(8, 4, 4, 2) + 0.1) # 调整边距适应长标签
# 创建箱线图
boxplot(data,
outline = FALSE, # 不显示异常值
notch = TRUE, # 显示中位数置信区间
las = 2, # 垂直x轴标签
col = "lightblue", # 添加颜色
main = ifelse(attr(data, "transform") == "log2",
"Expression Data (log2 transformed)",
"Expression Data (raw)"),
ylab = "Expression Level",
...)
# 添加网格线便于阅读
grid(nx = NA, ny = NULL, lty = 2, col = "gray")
}
# 执行可视化
enhanced_boxplot(processed_data)2.数据标准化
根据箱线图观察,样本之间存在差异,需要进行数据标准化
使用limma包的normalizeBetweenArrays函数对表达矩阵进行标准化
##根据箱线图,样本之间存在差异,所以需要进行数据标准化
# 使用limma包的normalizeBetweenArrays函数对表达矩阵进行标准化
# 默认采用分位数标准化方法(quantile normalization),使各样本分布一致
# 适用于消除技术变异,保留生物差异
exprSet = normalizeBetweenArrays(exprSet)
# 绘制标准化后的箱线图,验证标准化效果:
boxplot(exprSet,outline=FALSE, notch=T, las=2,
boxwex = 0.6, col = "orange") #- outline=FALSE: 不显示异常值点,图形更清晰; - notch=TRUE: 在箱体上显示凹槽,表示中位数的置信区间 - las=2: 使x轴标签垂直显示,便于阅读样本名称; - boxwex=0.6: 控制箱体宽度为默认的60%,避免拥挤;- col="orange": 设置箱体颜色为橙色,提高可视化效果
#标准化后,数据为 [1] "matrix" "array" # 检查标准化后数据的类型,通常是矩阵(matrix)或数组(array)
class(exprSet)
#转为数据框(data.frame)格式
exprSet <- data.frame(exprSet) 3.读取探针文件,进行ID转换
经过以上操作,我们就得到了行为基因名,列为样本名的数据框。每个基因log2转换后的表达数值不会过百,0-20以内。如果数值过大,或者出现异常值,或者报错,需要重新处理。
有了清洁数据,分组后就可以进行任何想要的火山图、热图、富集图和差异表达图等。
##探针ID和基因名的转换
library(readr)
dim(exprSet)#检查维度
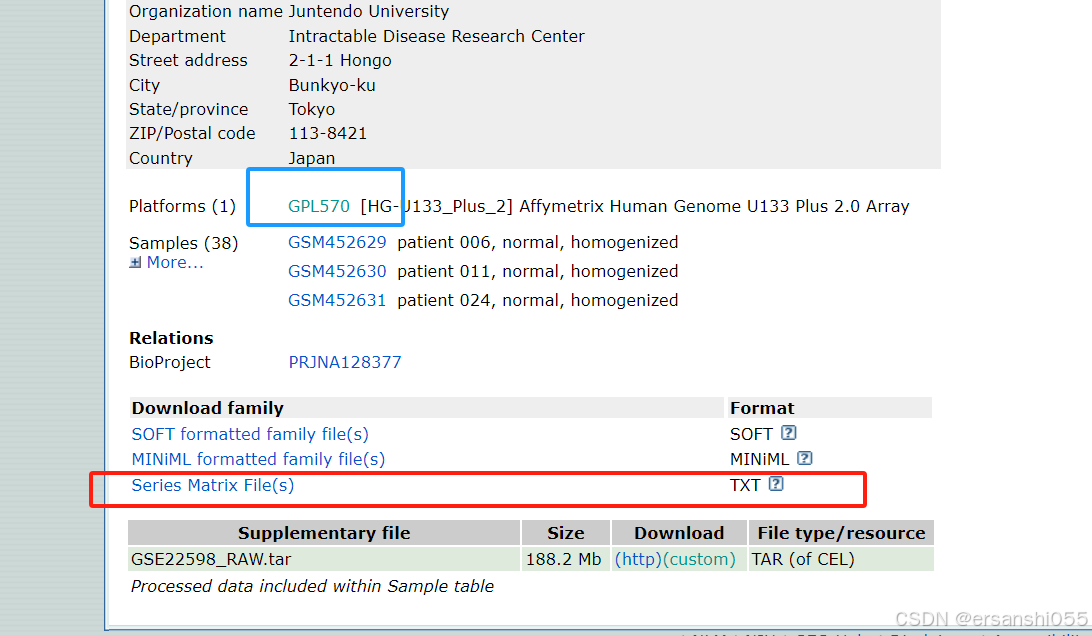
probe <- read_tsv("GPL570-55999.txt", skip = 16) #GEO探针格式为tsv,# 读取GPL570平台的探针注释文件(GEO数据库下载的tsv格式);skip = 16 跳过前16行文件头信息,直接读取数据部分
Probe_ID <- select(probe,c("ID","Gene Symbol"))# 从探针注释数据中提取需要的两列: - "ID":探针ID - "Gene Symbol":对应的基因符号
ids <- Probe_ID[-grep('///',Probe_ID$"Gene Symbol"),] #删除重复,数据清洗:删除基因符号不唯一的探针(包含"///"的条目表示映射到多个基因)
##将表达矩阵(exprSet)的行名(探针ID)转换为数据框中的显式列,方便后续与探针注释信息进行合并
library(dplyr)
library(tidyverse)
exprSet1 <- exprSet %>%
rownames_to_column("ID") #高频操作;
##使用内连接(inner_join)合并表达数据和探针注释信息,只保留在两个数据集中都存在的探针ID
exprset1 <- inner_join(ids,exprSet1,by="ID") #inner_join确保只保留有注释信息的探针
# length(exprset1$ID)# 检查探针数量
# length(exprset1$'Gene Symbol')# 检查基因符号数量
## 使用limma包的avereps函数对重复基因取平均值:
exprset1 = avereps(exprset1[,-c(1,2)], #高频操作
ID = exprset1$'Gene Symbol')#- exprset1[,-c(1,2)]:排除前两列(ID和Gene Symbol)的表达数据;- ID = exprset1$'Gene Symbol':指定基因符号作为分组依据;功能:将映射到同一基因的多个探针表达值取平均
dim(exprset1)#检查维度
## 将处理后的数据转换为数据框格式
exprset1 <- data.frame(exprset1) #获得行是基因名称,列是样本名称的表达矩阵(清洁数据)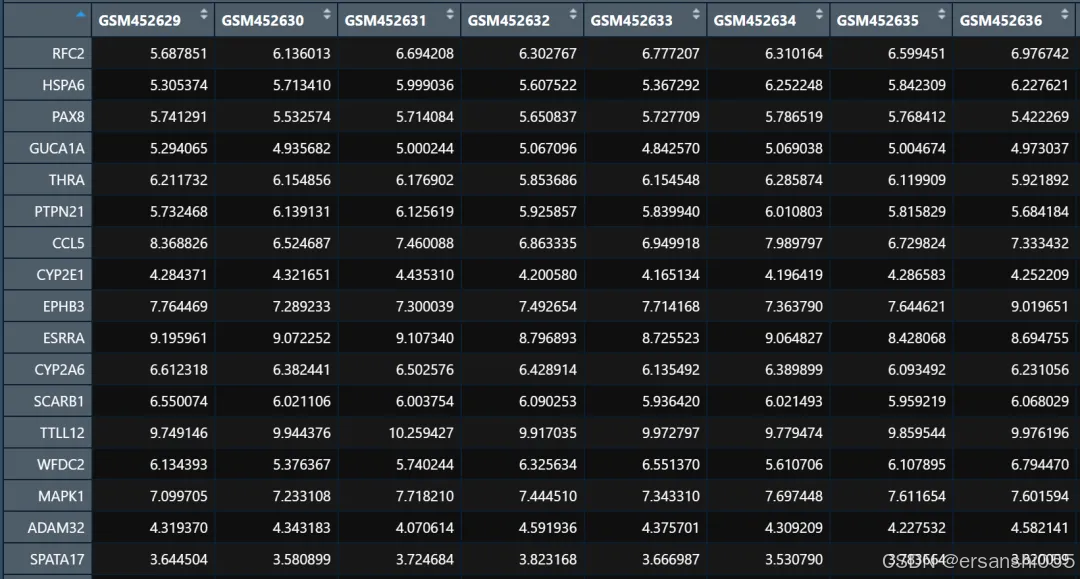
三、样本分组与差异分析
在GEO数据处理的过程中,样本分组往往决定差异的基因以及后续的各种分析。需要我们:①读懂研究设计,这个可以从GSE序列页面找;②能够实战操作,也就是在技能层面搞定。
例:以GSE22598为例,GEO Accession viewer,可分为三组,先舍弃cell line。这项研究是针对结肠癌的,样本包括两部分,一部分是17对临床样本,17个正常样本,17个肿瘤样本;还有加药处理的肿瘤细胞系,HT29和HCT15各1对,药物为表观遗传药物。数据来自 Clinical significance of UNC5B expression in colorectal cancer。
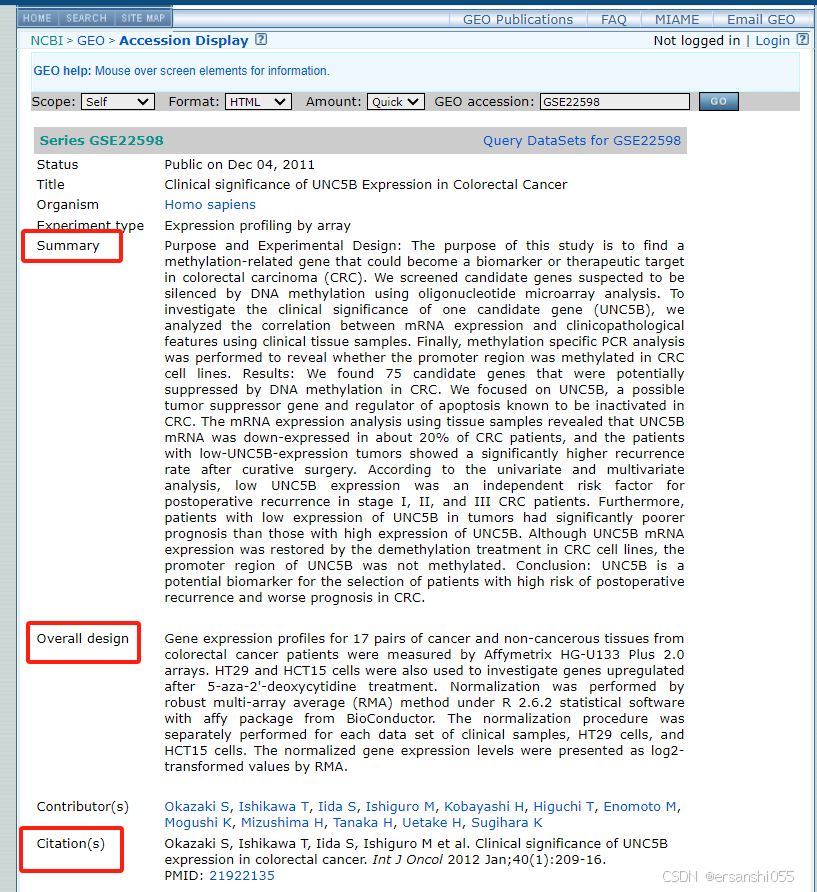
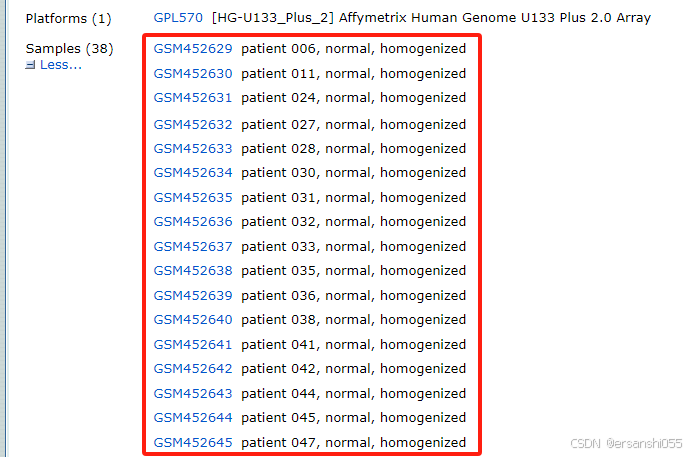
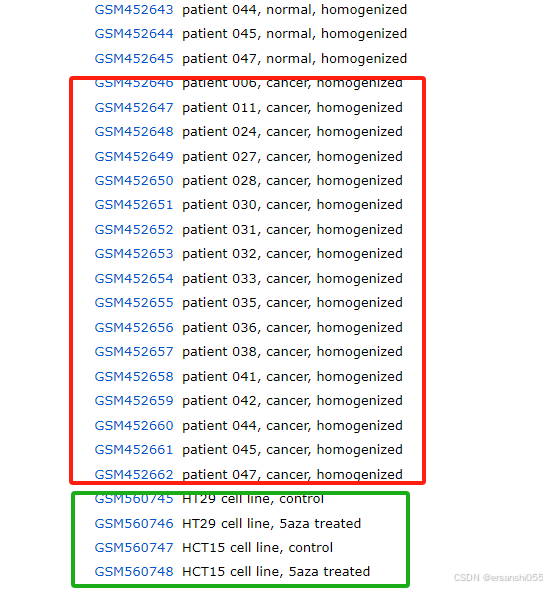
1.分组
(1)两组分组
我们选取前34个临床样本进行分析
########--------样本分组操作(以及差异分析)--------########
## 样本分组
exprset2 <- exprset1[,1:34]## 从表达矩阵中提取前34列样本数据(假设1-17是正常组,18-34是癌症组) 注意:实际应用中应根据实验设计确定分组方式
group1 <- c(rep("normal",17),rep("cancer",17)) ## 使用rep()函数重复创建分组标签
group1 <- factor(group1,levels = c("normal","cancer"))
# 将分组向量转换为因子(factor)并设置水平顺序:- levels参数指定"normal"为第一水平(将作为差异分析的参考组);- 因子化确保统计分析中正确处理分组变量
# 验证分组正确性
stopifnot(ncol(exprset2) == length(group1))
# 检查样本分布
print(table(group1))(2)多组分组
与两样本相比,多样本分组稍有差别。我们以GSE7476数据集为例。根据GSE7476数据集的实验设计,我们可以把样本分成两组:对照组和肿瘤组;也可以分为四组,即对照组、低级别组、中级别组和高级别组。
## 多组:样本分组
group2 <- c(rep("control",3),rep("low",3),rep("middle",3),rep("high",3))
group2 <- factor(group2)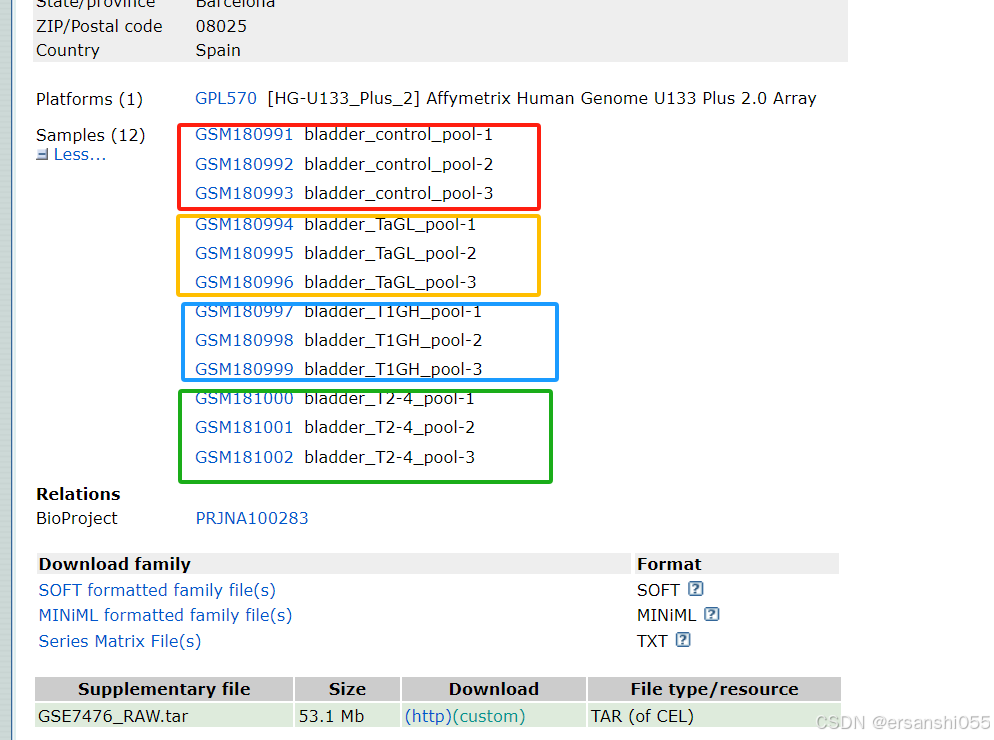
2.因子化
分组后,需要因子化才能进行拟合建模和差异分析。利用分组模型构建model.matrix ()和对比分析makeContrast ()。
(1)分组矩阵,model.matrix ()函数
在 R 语言中,mode1.matrix()函数可将一个数据框或公式对象转换为模型矩阵,以用于线性回归、生存分析等模型拟合。模型矩阵是一个二维矩阵,包含虚拟变量。虚拟变量,也叫哑变量,本质是将一个多分类变量转换为多个二分类变量,生成二分类变量的就是哑变量(如上图的理科类、文科类等)。虚拟变量是人为虚设的变量,通常取为0或1,以此反映某个变量的不同属性。
## 分组矩阵
# model.matrix(formula, data) #基本公式
design <- model.matrix(~0 + group1) #没有设置对照组
colnames(design) <- levels(group1)
design
#design <- model.matrix(~0 + group1)将样本分组信息转换为模型矩阵,并将其赋值给design变量。
## 比较矩阵
contrast.matrix <- makeContrasts( "cancer - normal", levels = design)(2)分组矩阵,makeContrast ()函数
对于多样本数据的分组,使用makeContrast ()函数。
介绍:
####---- makeContrasts()介绍----####
?makeContrasts()
makeContrasts(B-A,C-B,C-A,levels=c("A","B","C"))
## Levels B - A C - B C - A
A -1 0 -1
B 1 -1 0
C 0 1 1
makeContrasts(contrasts="A-(B+C)/2",levels=c("A","B","C"))
## Levels A-(B+C)/2
A 1.0
B -0.5
C -0.5
x <- c("B-A","C-B","C-A")
makeContrasts(contrasts=x,levels=c("A","B","C"))
## Levels B-A C-B C-A
A -1 0 -1
B 1 -1 0
C 0 1 1
3.通过拟合模型进行差异分析
lmFit()函数用于线性拟合模型构建,需要表达矩阵数据exprSet和分组矩阵design ,得到的结果再和对比矩阵contrast一起导入contrasts.fit()函数,转换为对比模型。在进行差异分析时,通过经验贝叶斯的方法进行评估,并通过topTable()函数获得差异分析结果。
## 差异表达分析流程
## 1.线性模型拟合
fit1 <- lmFit(exprset2,design)#使用limma包的lmFit函数拟合线性模型;- exprset2: 标准化后的表达矩阵(行是基因,列是样本); - design: 实验设计矩阵(之前创建的包含分组信息的矩阵)
## 2. 对比模型转换
fit1 <- contrasts.fit( fit1, contrast.matrix) # 转换为对比模型;- fit1: 已拟合的线性模型对象; - contrast.matrix: 之前定义的对比矩阵(如"cancer vs normal")
## 3. 经验贝叶斯评估
fit2 <- eBayes(fit1) # 经验贝叶斯评估;# 使用eBayes函数对标准误差进行经验贝叶斯调整;可以缩小基因间方差估计,提高差异检测的稳定性
## 4. 差异结果提取
allDiff = topTable(fit2,adjust='fdr',coef=1,number=Inf) # 差异分析结果提取芯片数据分析的流程已经标准化,形成了固有的模式,可以查看limma包。
四、可视化
在生信分析中,|log2FC| >1,p<0.05是生信分析的理想结果。在实验中,表达差异和表型验证,也要看到基因表达对表型的影响具有统计学差异。总之,生信分析的数据可视化的方式要根据待展示差异基因的数目而定。
差异基因较少(10^1数量级):柱状图、箱线图、散点图、小提琴图;
差异基因数多(10^2):热图
差异基因很多(10^3及以上):火山图
1.前期处理
差异分析的结果包括AveExpr(比较组间绝对值的平均差异值)、logFC(差异倍数)、t值、P值、q值(即adj.P.Val值)和B值。
t值,表示基因表达在不同样本之间差异的大小。P值,用于判断差异是否具有统计学上的显著性。q值,经过多重检验校正后的p值,用于控制假阳性的发生率。B值,是经过bayes调整后得到的标准差的对数值。一般logFC值、P值、q值和AveExpr值用来作为差异表达的判断标准,比如差异倍数在2倍以上、P值小于0.01等。根据基因、logFC、adj.P.Val或P.Value等关键信息即可绘制火山图和热图。
##保存差异基因的数据,临界值确定
diffSig <- allDiff[with(allDiff, (abs(logFC)>1 & adj.P.Val < 0.05 )), ]# 筛选显著差异基因(绝对logFC>1且FDR<0.05)
write.table(diffSig,file="diff.xls",sep="\t",quote=F)
diffUp <- allDiff[with(allDiff, (logFC>1 & adj.P.Val < 0.05 )), ]# 筛选上调基因(logFC>1且FDR<0.05)
write.table(diffUp,file="up.xls",sep="\t",quote=F)
diffDown <- allDiff[with(allDiff, (logFC<(-1) & adj.P.Val < 0.05 )), ]# 筛选下调基因(logFC<-1且FDR<0.05)
write.table(diffDown,file="down.xls",sep="\t",quote=F)
##数据整理和条件设置
data <- allDiff# 复制原始差异分析结果
data1 <- data %>%
rownames_to_column("Genes") #行名转为Genes为列名的一列
data2 <- data1 %>%
mutate(regulate = case_when(logFC >= 1 & adj.P.Val <= 0.05 ~ "up",
logFC <= -1 & adj.P.Val <= 0.05 ~ "down",
TRUE ~ "NS"))
#添加调控方向分类列, # 上调基因条件:logFC≥1且FDR≤0.05 ;下调基因条件:logFC≤-1且FDR≤0.05 ; 其他情况标记为无显著差异(Not Significant)
对于RNA-seq数据,可能考虑添加表达量过滤(如AveExpr>5)分类结果可用于后续火山图、热图等可视化
## 使用ggplot2包绘制火山图
# 基本绘图
library(ggplot2)
ggplot(data2,aes(logFC,-log10(adj.P.Val)))+
geom_point()+
labs(x=expression(Log[2]*" Fold Change"),
y=expression(-Log[10]*" (p value)")) #修改坐标轴命名细节
# 美化绘图
ggplot(data2,aes(logFC,-log10(adj.P.Val), #分别给正负显著变化的基因在图中根据颜色、大小标注出来
color=factor(regulate),
size=factor(regulate)))+
geom_point()+
labs(x=expression(Log[2]*" Fold Change"),
y=expression(-Log[10]*" (p value)"))+
theme_grey(base_size = 15)+
scale_color_manual(values = c('blue','grey','red'))+
scale_size_manual(values = c(2,1,2))+
theme(legend.title = element_blank(), #图例的设置参数
legend.position = "right", #标签位置为right
legend.background = element_rect(fill='transparent'))
# 增加注释基因
library(ggrepel)
# 筛选出ToP基因,并形成新列
data2$selectedgene <- ifelse(data2$adj.P.Val < 0.0001 & abs(data2$logFC) > 3,data2$Genes,NA)
ggplot(data2,aes(logFC,-log10(adj.P.Val),
color=factor(regulate),
size=factor(regulate)))+
geom_point()+
labs(x=expression(Log[2]*" Fold Change"),
y=expression(-Log[10]*" (p value)"))+
theme_grey(base_size = 15)+
scale_color_manual(values = c('skyblue','grey','tomato'))+
scale_size_manual(values = c(2,1,2))+
theme(legend.title = element_blank(),
legend.position = "right",
legend.background = element_rect(fill='transparent'))+
#用ggrepel包给选择的基因加上文本标签
geom_text_repel(aes(label=selectedgene), color="black",size=3,
box.padding=unit(0.5, "lines"),
point.padding=NA,
segment.colour = "black")
# 增加辅助线
data2$selectedgene <- ifelse(data2$adj.P.Val < 0.0001 & abs(data2$logFC) > 3,data2$Genes,NA)
ggplot(data2,aes(logFC,-log10(adj.P.Val),
color=factor(regulate),
size=factor(regulate)))+
geom_point()+
labs(x=expression(Log[2]*" Fold Change"),
y=expression(-Log[10]*" (p value)"))+
theme_grey(base_size = 15)+
scale_color_manual(values = c('skyblue','grey','tomato'))+
scale_size_manual(values = c(2,1,2))+
theme(legend.title = element_blank(),
legend.position = "right",
legend.background = element_rect(fill='transparent'))+
#用ggrepel包给选择的基因加上文本标签
geom_text_repel(aes(label=selectedgene), color="black",size=3,
box.padding=unit(0.5, "lines"),
point.padding=NA,
segment.colour = "black") +
geom_hline(yintercept = -log10(0.0001),linetype=2,cex=1)+ #添加辅助线
geom_vline(xintercept = c(-1,1),linetype=2,cex=1)+
annotate("text",x=-1.9,y=3.8,label="p value=0.0001",size=3)+ #添加一个注明
theme(axis.text= element_text(colour = "black"),
panel.border = element_rect(size=1,fill='transparent'))2.绘图,调整细节。
(1)火山图
## 使用ggvolcano包绘制火山图
# install.packages("devtools")
# devtools::install_github("BioSenior/ggVolcano")
library(ggVolcano)
ggvolcano(data = data2,x = "logFC",y = "adj.P.Val",output = FALSE,label = "Genes",
fills = c("#00AFFF", "#555555", "#FC4E07"),
colors = c("#00AFFF", "#555555", "#FC4E07"),
x_lab = "log2FC",
y_lab = "-Log10P.Value",
legend_position = "right") (2)热图
## 使用pheatmap绘制热图
library(pheatmap)
library(viridisLite)
pheatmap(heatdata)
heatdata <- exprset2[rownames(diffSig),]
annotation_col <- data.frame(group1) ##创建列的注释数据框
rownames(annotation_col) <- colnames(heatdata) ##行名换为样本名
pheatmap::pheatmap(heatdata, #热图数据
cluster_rows = F, #行聚类
cluster_cols =F, #列聚类,可以看出样本之间的区分度
annotation_col =annotation_col,
show_colnames=F,
scale = "row", #以行来标准化,这个功能很不错
color =colorRampPalette(c("blue", "white","red"))(10))
##美化热图
pheatmap(heatdata,
cluster_rows = TRUE,
cluster_cols = TRUE,
annotation_col =annotation_col,
annotation_legend=TRUE,
show_rownames = F,
scale = "row",
color = viridis(100, alpha = 1, begin = 0, end = 1, direction = 1),
cellwidth = 10,
cellheight = 0.2,
fontsize = 10)
##上调基因
topexp = exprset2[rownames(diffUp)[1:30],]
annotation_col = data.frame(group = group1)
rownames(annotation_col) = colnames(exprset2)
pheatmap(topexp,annotation_col = annotation_col,
color = colorRampPalette(c("blue", "gray", "red"))(10),
cellwidth = 10,
cellheight = 8,
fontsize = 8)五、富集分析(GO、KEGG)
1. 富集分析:包加载及数据读取
########--------富集分析-------########
##GO富集分析
library(readxl)
library(clusterProfiler)
library(ggplot2)
library(enrichplot)
library(GOplot)
library(DOSE)
library(stringr)
##读取数据或直接使用前面分析的数据data1
Data <- read_excel("diff.xls",sheet=1,na="NA") ##简单操作一下即可
gene <- bitr(Data$Genes,fromType = 'SYMBOL',toType = 'ENTREZID',OrgDb = 'org.Hs.eg.db') #基因名ID转换,把基因名转换成ENTREZID2.GO富集分析及绘图
## GO富集分析
GO<-enrichGO(
gene$ENTREZID,
OrgDb = 'org.Hs.eg.db',
keyType = "ENTREZID",
ont = "ALL",
pvalueCutoff = 0.01,
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
minGSSize = 50,
maxGSSize = 500,
readable = TRUE)
## GO富集分析绘图
barplot(GO, split="ONTOLOGY")+facet_grid(ONTOLOGY~., scale="free") +
scale_y_discrete(labels = function(x) stringr::str_wrap(x, width = 80)) ##避免字体重合
dotplot(GO, split="ONTOLOGY")+facet_grid(ONTOLOGY~., scale="free") +
scale_y_discrete(labels = function(x) stringr::str_wrap(x, width = 80))3.KEGG分析及绘图
##KEGG分析
KEGG <- enrichKEGG(
gene$ENTREZID,
organism = "hsa",
keyType = "kegg",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
minGSSize = 10,
maxGSSize = 500,
qvalueCutoff = 0.05,
use_internal_data = FALSE)
## KEGG富集分析绘图
barplot(KEGG,showCategory = 30,title = 'KEGG Pathway') +
scale_y_discrete(labels = function(x) stringr::str_wrap(x, width = 30))
dotplot(KEGG,showCategory = 30,title = 'KEGG Pathway') +
scale_y_discrete(labels = function(x) stringr::str_wrap(x, width = 30)) 可视化信号通路
## 绘制通路图
library("pathview")
gene$ENTREZID <- as.numeric(gene$ENTREZID )
pathview(gene.data = gene$ENTREZID,
pathway.id = "hsa04110",
species = "hsa",
out.suffix = "hsa04110.2layer",
kegg.native = T, same.layer = F)注意:无论是网站分析,还是 用 R 语言的分析结果,都需要实验来验证。
本文引用及参考资料:
不懂R,如何进行GEO数据库表达谱的差异分析、富集分析、蛋白互作、可视化?
ggplot2高效实用指南 (可视化脚本、工具、套路、配色)
https://zhuanlan.zhihu.com/p/210237979
The Gene Ontology Consortium. (2017). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research, 45(D1), D331–D338. https://doi.org/10.1093/nar/gkw1108.
PS:前期数据处理参考了芒果师兄的R 语言21天教程,致敬坚持的你!,有部分改动,并且添加了我自己在实战中理解的小技巧。关于R的操作,可以看我前边的博文或者生信宝典和生信技能树。

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐


 28
28 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)