单细胞Marker计算与可视化的pseudo-bulk方案
·
一、写在前面
pseudo-bulk:把同一个细胞类型/同一群细胞的多个单细胞表达量直接相加/求和,合成一个 “虚拟的bulk样本”。也许你会有疑惑,我们花这么多钱测得单细胞,不就是为了获得单细胞分辨率的转录组数据吗?为何还要将其再合并模拟生成bulk数据?
这其实为了解决单细胞稀疏性的问题,pseudo-bulk对scRNA-seq数据求和后,信号变强、噪声被平均掉。并且在scRNA-seq进行差异分析时,细胞之间不是独立样本,所以会干扰差异分析的准确度,从而产生假阳性。故差异基因更可靠、更接近真实生物学。
此前我们演示过pseudo bulk差异分析,接下来只需要转换一下变量,即可基于scRNA‑seq数据实现不同细胞类型间的pseudo bulk标记基因计算。
本教程基于Linux环境的rstudio演示,计算资源不足的同学可参考:
访问链接:https://biomamba.xiyoucloud.net/
欢迎联系客服微信[Biomamba_zhushou]获取帮助
如果需要单细胞数据分析教学、生信热点全文复现、自测数据个性化分析辅导、常态化实验学习,欢迎联系客服微信[Biomamba_zhushou]。
二、实操流程
1、数据基本情况
# 加载R包
library(Seurat)## Loading required package: SeuratObject## Loading required package: sp## 'SeuratObject' was built with package 'Matrix' 1.7.1 but the current
## version is 1.7.2; it is recomended that you reinstall 'SeuratObject' as
## the ABI for 'Matrix' may have changed##
## Attaching package: 'SeuratObject'## The following objects are masked from 'package:base':
##
## intersect, tlibrary (dplyr)##
## Attaching package: 'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union# 读取数据:
scRNA <- readRDS('test_data/T1D_scRNA.rds')
# 这个数据包含24个样本:
levels(scRNA$sample)## NULL# 包含两个组别的数据:
DimPlot(scRNA,split.by = 'Group')
# 取五种细胞类型用于下游演示:
select_ct <- paste0('celltype',1:5)
scRNA <- scRNA[,scRNA$cell_type %in% select_ct]
DimPlot(scRNA,split.by = 'Group')
2、循环添加细胞类型专属变量
for (run_ct in select_ct) {
target_col <- paste0(run_ct,'_var')# 细胞类型专属变量的slot名称
scRNA@meta.data[,target_col] <- scRNA$cell_type# 复制原始细胞变量
scRNA@meta.data[[target_col]][scRNA@meta.data[[target_col]] %>% as.character() != run_ct] <- 'OtherS' # 除了目标细胞类型外,其它的都叫OtherS
}3、循环生成pseudo-bulk数据并差异计算
3.1 生成拟bulk对象
bulk <- AggregateExpression(scRNA, return.seurat = T, slot = "counts", assays = "RNA",
group.by = c("cell_type", "sample", "Group",paste0(select_ct,'_var'))# 分别填写细胞类型、样本变量、分组变量的slot名称
)## Names of identity class contain underscores ('_'), replacing with dashes ('-')
## Centering and scaling data matrix
##
## This message is displayed once every 8 hours.# 生成的是一个新的Seurat对象:
bulk## An object of class Seurat
## 41056 features across 120 samples within 1 assay
## Active assay: RNA (41056 features, 0 variable features)
## 3 layers present: counts, data, scale.data3.2 pseudo-bulk计算细胞marker
# 差异分析会用到bulk分析常用到的DESeq2:
if(!require(DESeq2))BiocManager::install('DESeq2')## Loading required package: DESeq2## Loading required package: S4Vectors## Loading required package: stats4## Loading required package: BiocGenerics##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:dplyr':
##
## combine, intersect, setdiff, union## The following object is masked from 'package:SeuratObject':
##
## intersect## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
## match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
## Position, rank, rbind, Reduce, rownames, sapply, saveRDS, setdiff,
## table, tapply, union, unique, unsplit, which.max, which.min##
## Attaching package: 'S4Vectors'## The following objects are masked from 'package:dplyr':
##
## first, rename## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unname## Loading required package: IRanges##
## Attaching package: 'IRanges'## The following objects are masked from 'package:dplyr':
##
## collapse, desc, slice## The following object is masked from 'package:sp':
##
## %over%## Loading required package: GenomicRanges## Loading required package: GenomeInfoDb## Loading required package: SummarizedExperiment## Loading required package: MatrixGenerics## Loading required package: matrixStats##
## Attaching package: 'matrixStats'## The following object is masked from 'package:dplyr':
##
## count##
## Attaching package: 'MatrixGenerics'## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## Attaching package: 'Biobase'## The following object is masked from 'package:MatrixGenerics':
##
## rowMedians## The following objects are masked from 'package:matrixStats':
##
## anyMissing, rowMedians##
## Attaching package: 'SummarizedExperiment'## The following object is masked from 'package:Seurat':
##
## Assays## The following object is masked from 'package:SeuratObject':
##
## Assays# 创建空的数据框
mk_df <- data.frame()
for (run_ct in select_ct) {
# 改变默认分类变量:
Idents(bulk) <- paste0(run_ct,'_var')
# 下面的计算依赖DESeq2,做过Bulk RNA-Seq的同学都知道:
bulk_deg <- FindMarkers(bulk, ident.1 = run_ct, ident.2 = "OtherS", # 这样算出来的Fold Change就是run_ct/OtherS
slot = "counts", test.use = "DESeq2",# 这里可以选择其它算法
verbose = F# 关闭进度提示
)
bulk_deg$Celltype <- run_ct# 添加一列,收录当前进行差异计算的细胞类型
bulk_deg$gene <- rownames(bulk_deg)# 添加基因列
mk_df <- rbind(mk_df,bulk_deg)# 纵向合并数据框
}## converting counts to integer mode## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## converting counts to integer mode## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## converting counts to integer mode## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## converting counts to integer mode## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## converting counts to integer mode## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates# 计算top marker
top3_mk <- mk_df %>% group_by(Celltype) %>% top_n(n = 3, wt = avg_log2FC)
top3_mk## # A tibble: 15 × 7
## # Groups: Celltype [5]
## p_val avg_log2FC pct.1 pct.2 p_val_adj Celltype gene
## <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <chr>
## 1 6.52e- 37 3.45 1 0.823 2.68e- 32 celltype1 ENSG00000227240
## 2 3.00e- 20 3.89 0.958 0.25 1.23e- 15 celltype1 ENSG00000041515
## 3 3.25e- 14 3.87 0.958 0.281 1.33e- 9 celltype1 ENSG00000163629
## 4 1.74e- 65 4.50 1 0.396 7.15e- 61 celltype2 ENSG00000156395
## 5 8.25e- 53 4.10 1 0.177 3.39e- 48 celltype2 ENSG00000238241
## 6 3.55e- 16 4.22 0.875 0.25 1.46e- 11 celltype2 ENSG00000158966
## 7 0 7.18 1 0.615 0 celltype3 ENSG00000144645
## 8 0 7.40 1 0.625 0 celltype3 ENSG00000196092
## 9 5.37e-116 7.18 1 0.156 2.20e-111 celltype3 ENSG00000150625
## 10 3.34e- 18 4.05 1 0.604 1.37e- 13 celltype4 ENSG00000170624
## 11 4.24e- 18 4.05 0.833 0.333 1.74e- 13 celltype4 ENSG00000276241
## 12 9.19e- 10 3.61 0.667 0.073 3.77e- 5 celltype4 ENSG00000244649
## 13 8.73e-126 6.45 1 0.229 3.58e-121 celltype5 ENSG00000165061
## 14 1.17e- 57 5.45 0.958 0.229 4.80e- 53 celltype5 ENSG00000169744
## 15 2.17e- 35 5.40 1 0.427 8.91e- 31 celltype5 ENSG00000149403# 更改默认标签为细胞类型
Idents(bulk) <- bulk$cell_type
# 小提琴图看看我们拟bulk获得的基因效果如何:
VlnPlot(bulk,top3_mk$gene,stack = T)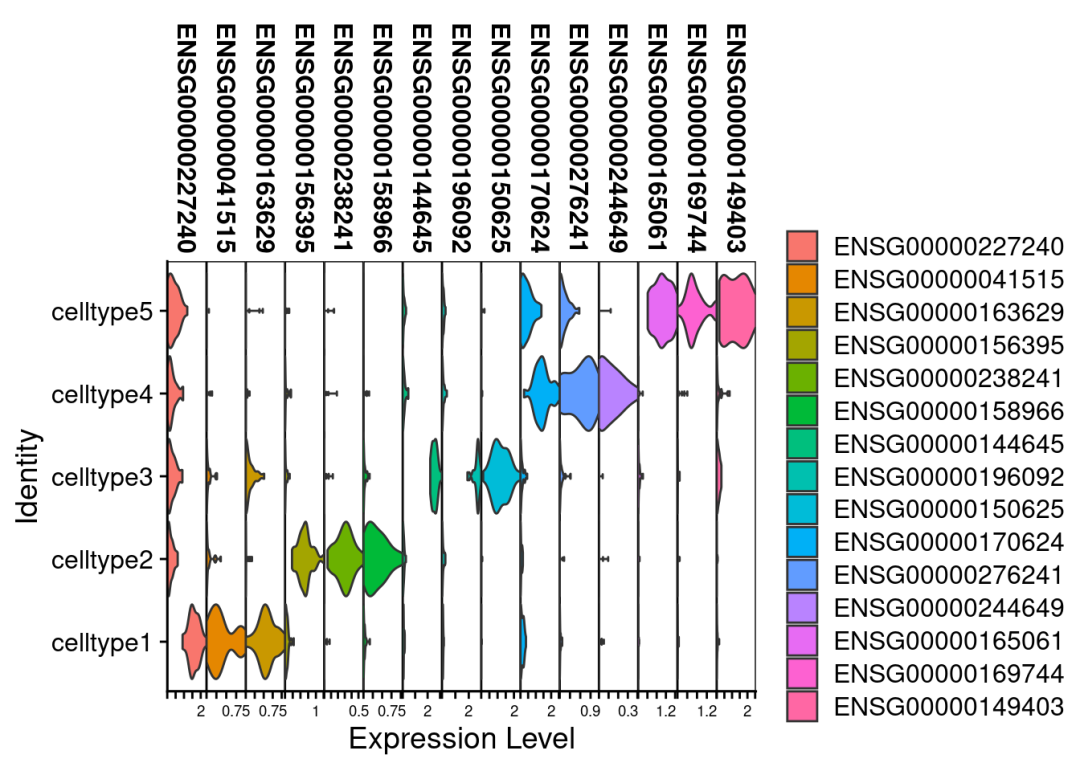
4、热图绘制
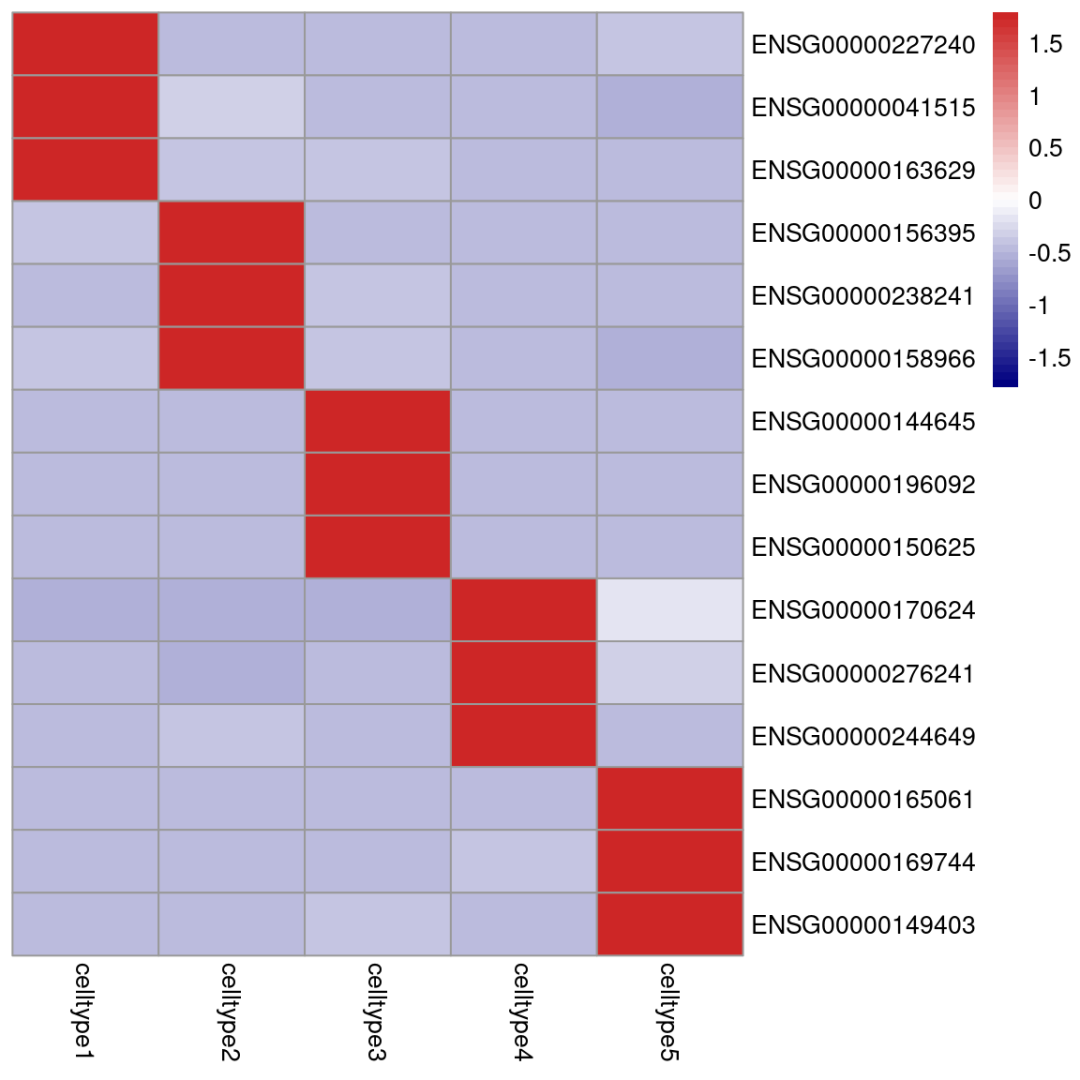
为了更有拟bulk的味道,热图也可以用平均值来绘制:
#获得表达均值矩阵:
avg <- AverageExpression(scRNA,features = top3_mk$gene)[["RNA"]]## As of Seurat v5, we recommend using AggregateExpression to perform pseudo-bulk analysis.
## This message is displayed once per session.avg <- as.data.frame(avg)
library(pheatmap)
# 绘制热图
pheatmap(avg[,select_ct],
cluster_rows = F,cluster_cols = F,
#行和列的聚类都关掉
,方便我们按顺序观察结果
show_colnames = T,
#展示横坐标的细胞分群
color = colorRampPalette(c("navy", "white", "firebrick3"))(50),
#修改渐变色
scale = "row"
)
三、演示环境
一切不给测试文件和分析教程都是耍流氓,本推送的代码和测试文件可以在以下链接中下载:

通过网盘分享的文件:
链接: https://pan.baidu.com/s/1RHUJksXVSHQKn8orPbiQmw
提取码: yxr9

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐

 7
7 0
0- 0



 已为社区贡献6条内容
已为社区贡献6条内容

所有评论(0)