DFT学习记录[2]:VASP能带计算全流程
本来想先写material project的使用方法,结果写了一下感觉完全不如先搞VASP的基本使用流程,所以这篇文章先讲一下VASP的基本使用全流程,从数据获取走到最后生成能带图,走完一遍之后就能大概理解vasp的使用流程了
如果有什么错误,请各位大佬直接指出来,这些东西都是我自己从众多项目和论文总结出来的比较直观的流程,如果有问题请直接指出来,谢谢您
1. 数据获取(material project)
数据就无脑用material project就好,不用问为什么,现在直接去官网下载即可,后续如果需要批量工作,会需要使用到api,这个就留到未来将mp的时候再说。
先进入网站
Materials Project![]() https://next-gen.materialsproject.org/materials/
https://next-gen.materialsproject.org/materials/
进入后注册登录,随便用什么注册都行(可能需要梯子)
然后你需要什么就在搜索栏里搜索就好了,或者点击元素周期表,比如我要AlN,那我就依次点击铝和氮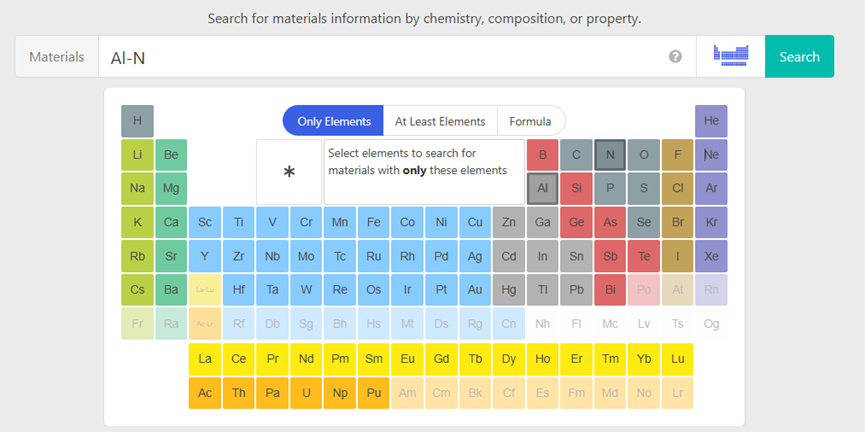
然后就能找到想要的AlN的各种物质了,我们使用mp-661,点击进入,可以在里面看到一些最基本的信息,你可以选择导出CIF,也可以导出POSCAR或者VASP input,如果导出CIF能够使用VESTA或者MS自己进一步检查并进行修改 ,用另外两个就少了自己建模的步骤,根据自己需求来吧
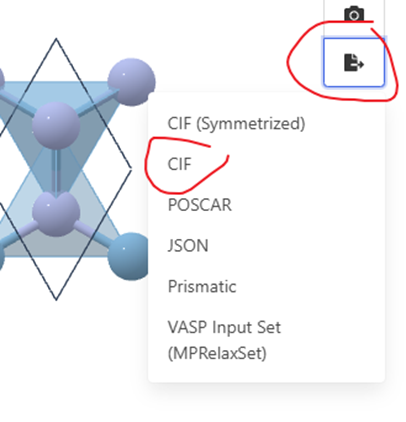
使用VESTA打开CIF文件可看到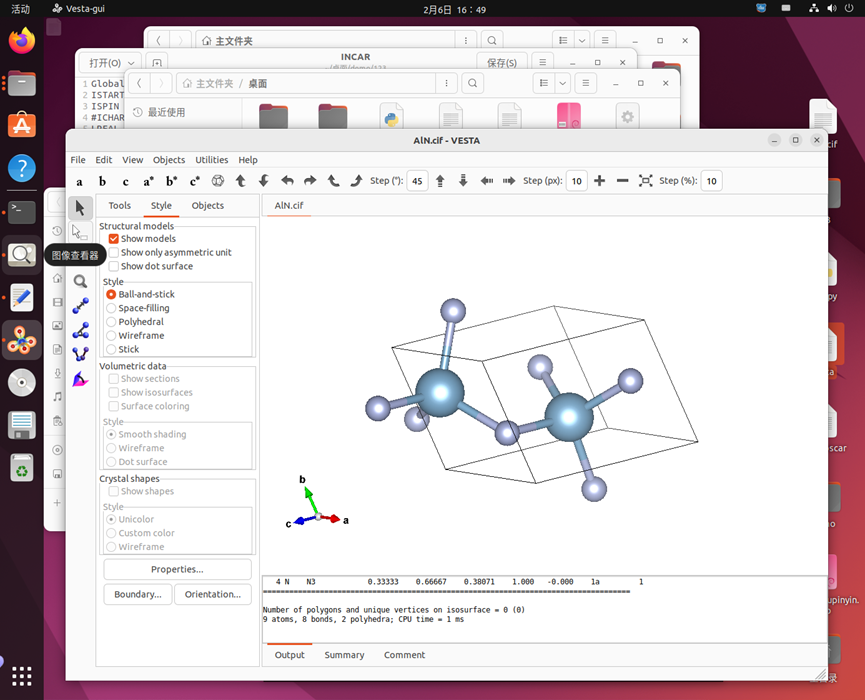
然后再左上角file选择export data,扔到文件夹中,这边为opt,然后把这个文件重名为POSCAR,这样就实现了和刚才获取数据时没有选择的POSCAR文件,这个是进行VASP运算的核心文件之一
2.VASPkit预处理
完成之后命令台进入该文件夹,输入vaspkit来启动vaspkit
然后使用1选择VASP Input-Files Kit
然后在这里面生成我们进行VASP计算需要的其他几个文件,分别为INCAR,KPOINTS,POTCAR
直接输入101选择Customize INCAR File ,然后使用SR选择标准弛豫,这样就生成INCAR文件了,具体流程见下代码块
===================== Structural Utilities ======================
1) VASP Input-Files Kit 2) Mechanical Properties
3) K-Path for Band-Structure 4) Structure Editor
5) Catalysis-ElectroChem Kit 6) Symmetry Analysis
8) Advanced Structure Models
===================== Electronic Utilities ======================
11) Density-of-States 21) Band-Structure
23) 3D Band-Structure 25) Hybrid-DFT Band-Structure
26) Fermi-Surface 28) Band-Structure Unfolding
31) Charge-Density Analysis 42) Potential Analysis
51) Wave-Function Analysis 62) Magnetic Properties
65) Spin-Texture 68) Transport Properties
======================== Misc Utilities =========================
71) Optical Properties 72) Molecular-Dynamics Kit
74) User Interface 78) VASP2other Interface
91) Semiconductor Kit 92) 2D-Material Kit
0) Quit
------------>>
1
==================== VASP Input Files Options ===================
101) Customize INCAR File
102) Generate KPOINTS File for SCF Calculation
103) Generate POTCAR File with Default Setting
104) Generate POTCAR File with User Specified Potential
105) Generate POSCAR File from cif (no fractional occupations)
106) Generate POSCAR File from Material Studio xsd (retain fixes)
107) Reformat POSCAR File in Specified Order of Elements
108) Successive Procedure to Generate VASP Files and Check
109) Submit Job Queue
0) Quit
9) Back
------------>>
101
+-------------------------- Warm Tips --------------------------+
You MUST Know What You Are doing
Some Parameters in INCAR File Neet To Be Set/Adjusted Manually
+---------------------------------------------------------------+
======================== INCAR Options ==========================
ST) Static-Calculation SR) Standard Relaxation
MG) Magnetic Properties SO) Spin-Orbit Coupling
D3) DFT-D3 no-damping Correction H6) HSE06 Calculation
PU) DFT+U Calculation MD) Molecular Dynamics
GW) GW0 Calculation BS) BSE Calculation
DC) Elastic Constant EL) ELF Calculation
BD) Bader Charge Analysis OP) Optical Properties
EC) Static Dielectric Constant PC) Decomposed Charge Density
FD) Phonon-Finite-Displacement DT) Phonon-DFPT
NE) Nudged Elastic Band (NEB) DM) The Dimer Method
FQ) Frequence Calculations LR) Lattice Relaxation
0) Quit
9) Back
------------>>
Input Key-Parameters (STH6D3 means HSE06-D3 Static-Calcualtion)
SR
-->> (01) Written INCAR file!
+---------------------------------------------------------------+
| * ACKNOWLEDGMENTS * |
| Other Contributors (in no particular order): Peng-Fei LIU, |
| Xue-Fei LIU, Dao-Xiong WU, Zhao-Fu ZHANG, Tian WANG, Qiang LI,|
| Ya-Chao LIU, Jiang-Shan ZHAO, Qi-Jing ZHENG, Yue QIU and You! |
| Advisors: Wen-Tong GENG, Yoshiyuki KAWAZOE |
| Any Suggestions for Improvement are Welcome and Appreciated! |
+---------------------------------------------------------------+
| * CITATIONS * |
| When using VASPKIT in your research PLEASE cite the paper: |
| [1] V. WANG, N. XU, J.-C. LIU, G. TANG, W.-T. GENG, VASPKIT: A|
| User-Friendly Interface Facilitating High-Throughput Computing|
| and Analysis Using VASP Code, Computer Physics Communications |
| 267, 108033, (2021), DOI: 10.1016/j.cpc.2021.108033 |
+---------------------------------------------------------------+然后生成KPOINTS,分别为1,102,1,最后这个是让你选择精度,如果你只是为了练手,那0.05即可,输入之后案例会将KPOINTS和POTCAR一起生成,但是如果POTCAR没有生成则说明你没有正常设置vaspkit的赝势库,所以我们现在设置一下这个,先去这里把文件下载下来
VASP赝势库文件 - 哔哩哔哩![]() https://www.bilibili.com/read/cv23515006/?opus_fallback=1
https://www.bilibili.com/read/cv23515006/?opus_fallback=1
然后修改~/.vaspkit,可以用sudo gedit ~/.vaspkit
然后按照他的方法把这些路径改成你保存的路径
其中也有一个PYTHON_BIN,要把这个改成你的python路径,还有一个AUTO_PLOT,可以自动画图,也该成TRUE,完成之后重新跑一遍就能正常生成了。
3.VASP的正式运行
首先我们先说一下这几个文件的用处:
POSCAR最核心的文件,用来描述我们要研究的物质的结构
INCAR次要,用来设置我们每次计算的各种参数
POTCAR为赝势文件,描述原子电子间相互作用
KPOINTS你可以简单理解为我们计算的精度要求(与前面使用vaspkit让我们设置精度呼应)
是不是已经很麻烦了,后面还会更麻烦,我们使用VASP的时候,是需要分多次计算来的,每一次计算完成都需要对INCAR进行调整以进行下一次计算,拿能带计算举例子,通常需要这几个步骤
- opt(结构优化):先让原子位置和晶格参数弛豫到能量最低的稳定状态,避免用初始不合理的结构导致后续电子性质计算失真。
- scf(静态自洽计算):基于优化后的结构,精确求解电子密度和波函数,生成 CHGCAR、WAVECAR 等文件,为后续非自洽计算提供准确的电子态信息。
- band(能带计算):属于非自洽计算,沿着布里渊区的高对称 k 点路径计算电子能量,最终得到能带图,用于分析材料的电子输运、带隙等性质。
所以我们需要调整三次INCAR文件,至于INCAR文件该怎么设置,这是个大学问,是需要多次学习与长久的经验才能熟练掌握的,就像CS外挂的参数师,这是一个不错的INCAR各参数介绍,可以学习一下
那我们先进行opt结构优化的相关计算
3.1opt结构优化计算
首先打开INCAR,能发现其中的内容大致为
Global Parameters
ISTART = 1 (Read existing wavefunction, if there)
ISPIN = 1 (Non-Spin polarised DFT)
# ICHARG = 11 (Non-self-consistent: GGA/LDA band structures)
LREAL = .FALSE. (Projection operators: automatic)
# ENCUT = 400 (Cut-off energy for plane wave basis set, in eV)
# PREC = Accurate (Precision level: Normal or Accurate, set Accurate when perform structure lattice relaxation calculation)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid, helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells, be careful)
# LPLANE = .TRUE. (Real space distribution, supercells)
# NWRITE = 2 (Medium-level output)
# KPAR = 2 (Divides k-grid into separate groups)
# NGXF = 300 (FFT grid mesh density for nice charge/potential plots)
# NGYF = 300 (FFT grid mesh density for nice charge/potential plots)
# NGZF = 300 (FFT grid mesh density for nice charge/potential plots)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing, metals:1)
SIGMA = 0.05 (Smearing value in eV, metals:0.2)
NELM = 90 (Max electronic SCF steps)
NELMIN = 6 (Min electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence, in eV)
# GGA = PS (PBEsol exchange-correlation)
Ionic Relaxation
NSW = 100 (Max ionic steps)
IBRION = 2 (Algorithm: 0-MD, 1-Quasi-New, 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -2E-02 (Ionic convergence, eV/AA)
# ISYM = 2 (Symmetry: 0=none, 2=GGA, 3=hybrids)我们做个简单的调整,首先看到ENCUT

为了让结果更准确一点,我们最好还是要把ENCUT手动调整一下,去掉注释,我们要把ENCUT设置为默认值的1.3倍,所以调整为520
PREC可以调整计算的精度,你可以根据自己的需要调整,我们就不改了
接下来是LWAVE 和LCHARG,这两个是把计算的波函数保存下来,我们这一步用不到,所以可以改成FALSE
剩下的基本都是调整精度的了,我就不调了,把注释的删掉后留下(注:ICHARG后续会用到所以保留)
Global Parameters
ISTART = 1 (Read existing wavefunction, if there)
ISPIN = 1 (Non-Spin polarised DFT)
# ICHARG = 11 (Non-self-consistent: GGA/LDA band structures)
LREAL = .FALSE. (Projection operators: automatic)
ENCUT = 520 (Cut-off energy for plane wave basis set, in eV)
LWAVE = .FALSE. (Write WAVECAR or not)
LCHARG = .FALSE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid, helps GGA convergence)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing, metals:1)
SIGMA = 0.05 (Smearing value in eV, metals:0.2)
NELM = 90 (Max electronic SCF steps)
NELMIN = 6 (Min electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence, in eV)
Ionic Relaxation
NSW = 100 (Max ionic steps)
IBRION = 2 (Algorithm: 0-MD, 1-Quasi-New, 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -2E-02 (Ionic convergence, eV/AA)那我们现在INCAR就已经设置完毕了,现在可以进行运行了,想要运行vasp我们是需要脚本来实现的,我的脚本起名为run_vasp.sh
#!/bin/bash
BIN=/home/blinder/下载/vasp.6.3.2/bin/vasp_std
mpirun -np 8 $BIN > vasp.out第一行不用动, BIN为你的vasp_std的路径,-np8为你运行使用的核心数,并不一定是越多越好,一般个人电脑设置8就够,完成之后,进入到你放之前四个文件的opt文件夹中,使用下述代码即可运行,或者你可以输入.后空格,然后把文件拖进来也可以,然后就开始跑了
. run_vasp.sh如果你想实时看到当前的进度,可以开一个新的命令台,然后进入文件夹,输入
tail -f vasp.out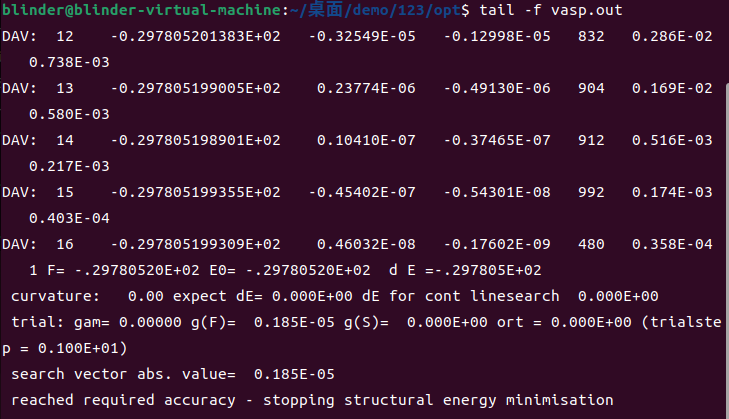
看到最后出现reached required accuracy - stopping structural energy minimisation即为结束
生成了一大堆文件,其中最重要的是CONTCAR,这是优化之后的结构,我们后续需要将其作为新的POSCAR进行后续的计算,你可以把他拖进VESTA或者MS进行检查
okay,我们发现已经没毛了,那我们就进行下一步
3.2 scf静态自洽计算
首先我们把CONTCAR INCAR KPOINTS POTCAR复制到新的scf文件夹,然后把CONTCAR重命名为POSCAR
接下来还是像之前一样,我们要调整INCAR


首先把NSW设置成1,然后把LWAVE和LCHARG设置成TRUE,我们第三步需要这两部分的信息,所以需要让它保存计算结果,其他的改不改都行,基本都只是精度问题
然后保存,继续进入该文件夹然后运行脚本运行,这次的结尾多了一个东西,就是在保存波函数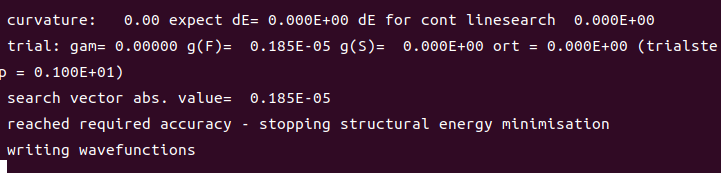
3.3 band能带计算
上次生成的文件最关键的如下图,我们把他放到新的文件夹band里,你会发现没有KPOINTS,因为我们原本生成的KPOINT在最后一步能带计算并不是很好用,所以我们需要额外生成一个,我们使用VASPKIT
进入 3) K-Path for Band-Structure ,这个时候他问我们我们的结构是几维的,之前通过VESTA能够清楚地看出来是三维的,我们就直接303) Bulk Structure ,按理来说会生成一个布里渊区的示意图,但是可能会出现报错
AttributeError: 'Arrow3D' object has no attribute 'do_3d_projection'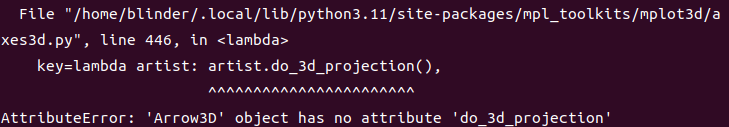
这个是因为我们使用的vaspkit的版本太老了,使用matplotlibs的函数很落后,所以这个时候需要对vaspkit进行修改,参考下链接
【新提醒】VASPkit绘制布里渊区图像出错 - VASPKIT使用相关 - VASPKIT论坛 - VASPKIT![]() http://bbs.vaspkit.cn/thread-374-1-1.html
http://bbs.vaspkit.cn/thread-374-1-1.html
但是我在按照这个帖子进行修改之后发现还是不行,继续研究后发现是matplotlib的版本太高了,所以可以直接
pip install matplotlib==3.4.3 -i https://pypi.tuna.tsinghua.edu.cn/simple/然后继续运行,发现又报错了
AttributeError: `np.Inf` was removed in the NumPy 2.0 release. Use `np.inf` instead.所以再跑一个
pip install numpy==1.26.4 -i https://pypi.tuna.tsinghua.edu.cn/simple/现在就正常了
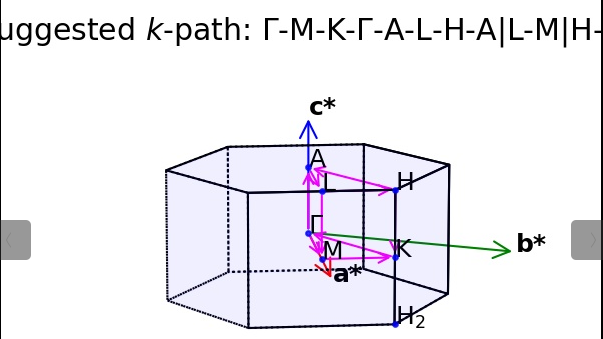
这个时候会生成一个KPATH.in,这个就是我们需要的新的KPOINTS,所以我们把他重命名
接下来要修改INCAR,首先把ICHARG = 11取消注释

然后LWAVE和LCHARG你可以根据自己的需求选择要不要取消保存,我这里就保存了,然后直接跑运行脚本
恭喜你,我们的vasp计算到此结束了,我们可以利用vaspkit把图画出来,仍然是在这个文件夹打开命令台,vaspkit,然后21,211,1,即可完成画图
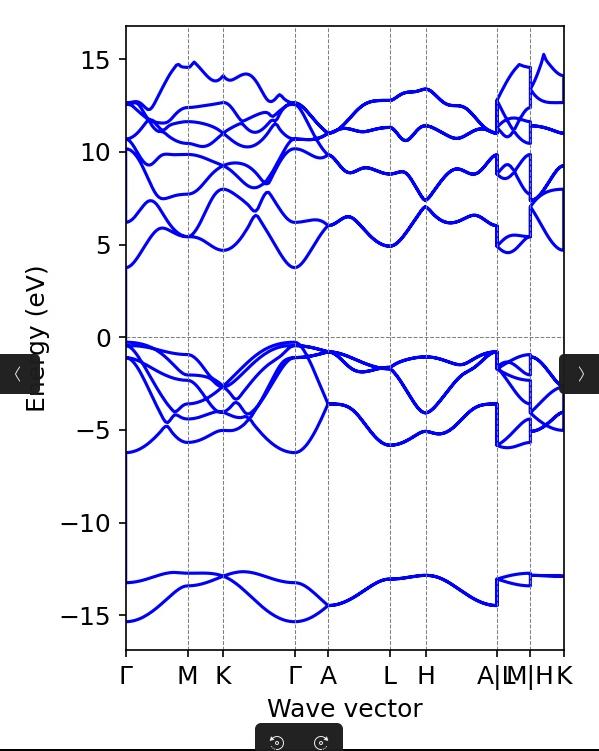
如果你觉得这个图比较丑,你想修改参数,就可以去PLOT.in里面进行画图参数的修改
文件夹中生成的.dat文件也可以直接扔到origin里面进行画图
你看BAND_GAP能看到,我们算出来的能带宽度为4.0238
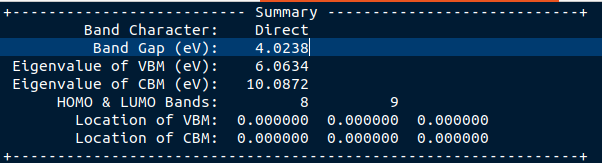
material project上为4.05
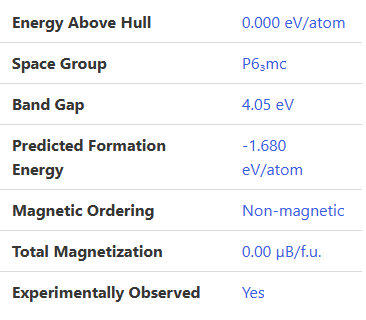
那说明我们的计算精度确实还不错
如果你去网页上或者文献里找,经常能看到说AlN的能带宽度为6左右,这是计算与实验难以避免的误差,问题不大,如果想要尽量减小这部分的误差,可以使用GW等更精确的计算方法,但是效率也会随之降低。相关解答可见该帖子
采用VASP计算得到的禁带宽度为什么比实验值低 - 第一性原理 - Vasp&MedeA - 小木虫论坛-学术科研互动平台![]() https://muchong.com/t-7486590-1
https://muchong.com/t-7486590-1
就是这样了,这就是完成一次能带计算的比较系统的流程,如果存在任何问题请指出来,我会及时解答或者修改,接下来应该会搞一下material project的api调用教程,敬请期待

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐



 16
16 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)