达摩院MICCAI'25|“空间蛋白”的神经辐射场:实现空间蛋白质组的精准预测
作者|赵舶凯,阿里巴巴达摩院实习生
引言
空间蛋白质组学技术通过解析蛋白质在组织中的分布特征,正在重塑生命科学研究的格局。然而,现有基于测序的技术受限于空间分辨率不足,难以精准刻画复杂组织结构特征。
为了实现空间分辨率的提升,当前计算方法通过数字病理图像特征预测未检测位点的分子信息。然而这些方法对分子空间位置信息的建模存在显著不足,导致无法有效预测蛋白质表达。
为了解决这一问题,本文首次提出高分辨率蛋白质空间表达预测框架Neural Proteomics Fields(NPF)。我们将空间组学的预测问题建模为连续空间中的蛋白质表达重建问题。通过对每个组织样本训练一个专门的网络用于蛋白表达预测。NPF包括一个空间建模模块,学习组织特异性蛋白质的空间分布,和一个形态建模模块,提取组织特异性的形态特征。
此外,本文基于多重荧光蛋白图像构建了基准数据集(pseudo-Visium SP),并补充真实数据 10X Visium CytAssist Protein Expression 以评估算法性能。实验结果表明,在较少可学习参数的情况下,NPF 能实现最先进的性能,证明了其在空间组学研究中的应用潜力。
论文链接:https://arxiv.org/abs/2508.17389
代码仓库:https://github.com/Bokai-Zhao/NPF

介绍
空间组学(spatial omics),包含了一系列能够在组织原始空间位置中测量其分子特征的技术,已被广泛视为生命科学领域的新前沿。
空间蛋白质组技术, 通过从组织解析蛋白质信息, 凭借其迅速的发展被《自然-方法》评选为2024年"年度方法",并推动肿瘤研究与组织图谱构建工作。

现有空间组学技术主要分为两大类:基于成像的技术和基于测序的技术。基于成像的技术使用金属粒子(IMC,MIBI)或荧光(CODEX)标记蛋白的方法,受限于金属粒子数量和成像通道数,所检测的蛋白质数量受限;基于测序的技术理论上可以实现高通量的蛋白质检测,但受限于测序位点的精度,空间分辨率受限。基于测序的空间蛋白组的空间分辨率受限于测序位点的间距和位点半径。因此,可以通过预测没有测序位点的蛋白质表达,提升测序方法的分辨率。
基于测序的空间蛋白组的空间分辨率受限于测序位点的间距和位点半径。因此,可以通过预测没有测序位点的蛋白质表达,提升测序方法的分辨率。经典的空间组学分子预测方法STNet、istar将 ST 预测问题视为传统的回归问题,其中网络以 WSI 补丁作为输入,并产生相应补丁区域内组织的基因表达强度。

目前的预测方法主要用于空间转录组,由于数据形式相同,可以直接用于空间蛋白表达的预测,然而仍存在以下问题:
-
生物分子具有空间分布特征,仅靠局部图像特征进行分子预测,没有考虑到蛋白质分布的空间位置关系;部分方法通过图神经网络建模测序位点之间的邻域关系,忽略了对WSI的全局空间建模。
-
蛋白分布在不同组织中存在分布差异,因此需要充分利用WSI中的图像特征,istar使用预训练的 HIPT 抽取图像特征,忽略了不同组织的异质性,STNet直接从组织补丁中训练深度神经网络,受限于训练的WSI中的补丁数量,难以从每个测序点图像中提取细粒度的特征。
-
目前没有对空间蛋白质组预测问题的benchmark,真实公开的数据集有限,使用真实的测序数据集无法有效对密集预测任务进行交叉验证。
本工作从两方面解决着手解决这些问题:
模型方面:
-
针对空间建模不足的问题,我们将空间蛋白质预测问题建模为连续空间上的蛋白质表达重建问题。受到计算机视觉中的三维重建技术启发,我们引入空间坐标编码模块。
-
针对组织特异性表征提取,我们设计了病理图像特征提取模块,将病理基础模型引入空间蛋白质预测任务。
数据方面:
-
我们通过高分辨率多重荧光成像的虚拟采样,生成构建了首个面向空间蛋白质组学的基准数据集,突破了真实蛋白质空间数据稀缺的局限。
-
额外使用真实数据外部验证,为领域内算法评估建立了标准化测试,解决了既往依赖单一数据集导致的评估偏差问题。
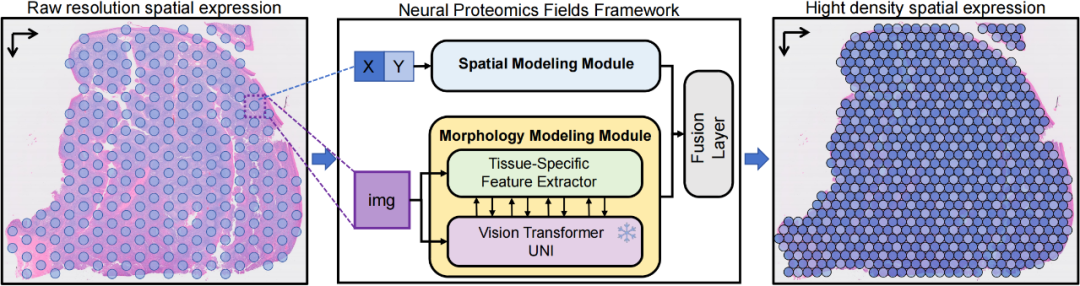

核心方法
问题建模:我们将空间蛋白质预测问题建模为连续空间上的蛋白质重建问题。
-
三维重建问题:单个物体 稀疏视角 -> 密集重建(3D);
-
空间组学预测:单张切片 稀疏检测 -> 密集生成(2D)。
受到计算机视觉中的三维重建技术Nerf启发,我们引入空间坐标编码模块,捕捉蛋白质表达随空间位置变化的细微梯度特征,构建空间感知架构。
此外,我们设计了病理图像特征提取模块,将病理基础模型引入空间蛋白质预测任务。在保留预训练模型强表征能力的同时,显著减少了可训练参数,解决了传统方法因组织异质性或数据量限制导致的特征提取瓶颈,实现了细粒度、多层次的病理图像表征。
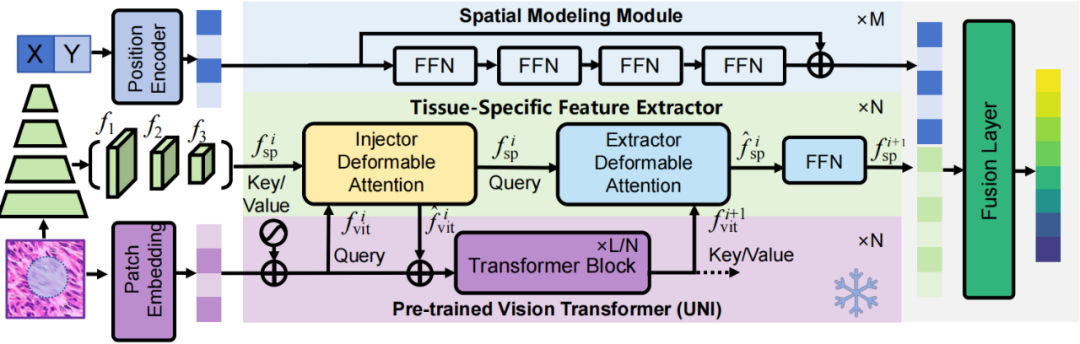
数据构建:我们通过高分辨率多重荧光成像的虚拟采样生成了虚拟的基于测序技术的数据集。
简单来说,我们通过对多重荧光蛋白图像与组织病理学图像配准得到配对数据,并在蛋白图像上进行虚拟采样点设置,根据采样点的物理半径计算虚拟的蛋白表达。通过间隔排布采样点构建了四个互不重叠的子集。该数据集模拟了测序技术的空间分布特性,支持交叉验证的严谨评估范式。


结果评估
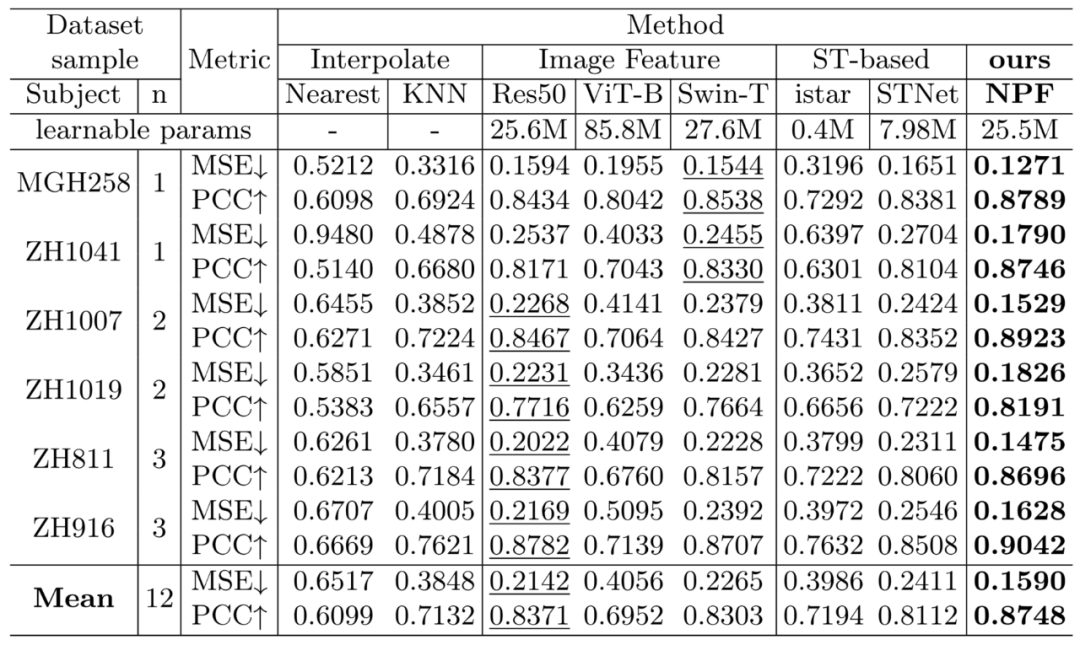
我们在构建的六个被试12张切片的数据集上进行了严格的交叉验证,并与插值方法、基于病理图像预测的方法和空间转录组的经典方法进行了对比,我们的方法更具优势。
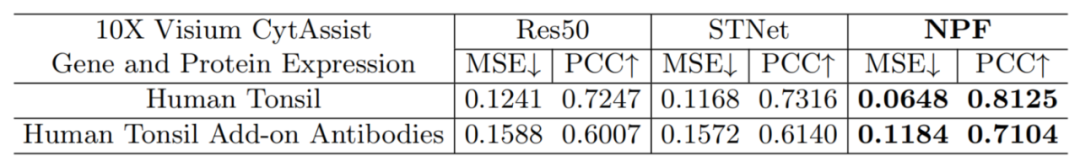
此外,我们在真实的10x数据集进行了验证,根据采样点进行7:1:2的train/valid/test多次随机数据划分,结果表明我们的方法在真实数据集上拥有更大的优势。
可视化结果表明,通过NPF生成的高分辨率的蛋白质组数据更接近真实的蛋白质分布:
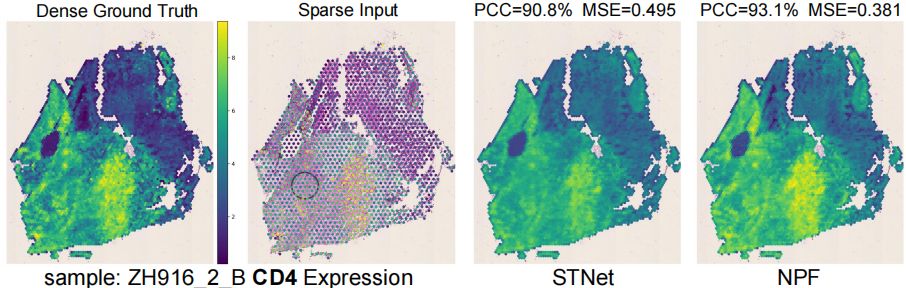
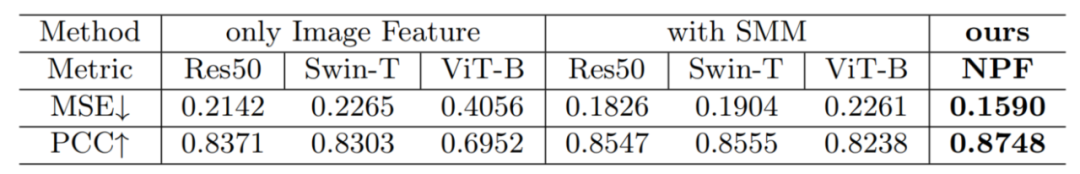
我们还发现将空间建模与其他图像编码器结合,能够提升蛋白质预测的精度,表明了空间建模方法在空间蛋白质组预测中的潜力。

结语
我们首次提出了一项针对测序空间蛋白质组进行空间超分辨率分析的新任务,并为此设计了一个名为NPF的神经辐射场框架,旨在通过全切片图像(WSI)预测空间蛋白质表达。
该框架集成了空间建模与形态学建模模块,为高通量蛋白质表达的精准预测提供了一条有效途径。在模拟及真实数据集上的广泛实验均表明,NPF的性能达到了业界领先水平。更重要的是,NPF为空间组学领域中的空间建模方法开创了新的道路。我们未来的研究方向包括:拓展空间建模的应用场景,并将其与其他空间组学数据融合,更深刻地揭示组织微环境的复杂性,用于图谱构建、癌症研究等领域。

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐

 0
0 0
0- 0



 已为社区贡献120条内容
已为社区贡献120条内容

所有评论(0)