lammps软件_高熵合金模拟计算(Npj. Comput. Mater)中简单的LAMMPS模拟
各位研究材料的同学都知道,现在高熵合金(HEAs)的研究很火,但几乎绝大部分是都是实验研究,很少看到有理论模拟方面的研究。不过我还是找到一篇,来自韩国浦项科技大学李秉珠 (Byeong-Joo Lee)领导的研究小组采用各种模拟技术对高熵合金的研究,这也是我看到的第一篇关于高熵合金模拟计算的文章。此研究成果发表在了Npj. Comput. Mater这个模拟计算类大神云集的期刊上。...
·

各位研究材料的同学都知道,现在高熵合金(HEAs)的研究很火,但几乎绝大部分是都是实验研究,很少看到有理论模拟方面的研究。不过我还是找到一篇,来自韩国浦项科技大学李秉珠 (Byeong-Joo Lee) 领导的研究小组采用各种模拟技术对高熵合金的研究,这也是我看到的第一篇关于高熵合金模拟计算的文章。此研究成果发表在了 Npj. Comput. Mater 这个模拟计算类大神云集的期刊上。简单点说就是他们的研究阐明了高熵合金中某些材料现象的物理冶金学原理(如在低温下的迟滞扩散和微孪晶的形成),并利用原子尺度的模拟(蒙特卡罗方法、分子动力学和分子静力学)研究了各元素对等原子比例的 CoCrFeMnNi 高熵合金固溶度的影响。以下是文章的题目:
 今天就通过这篇文章给大家介绍里面关于可以使用LAMMPS软件进行的一些简单的分子动力学模拟:●高熵合金模型的构建关于这个用 LAMMPS 软件构建高熵合金模型是我本人临时想到的,因为最近翻看 LAMMPS 手册时发现了一个很有意思的命令,我们先看一下文章中的高熵合金模型:
今天就通过这篇文章给大家介绍里面关于可以使用LAMMPS软件进行的一些简单的分子动力学模拟:●高熵合金模型的构建关于这个用 LAMMPS 软件构建高熵合金模型是我本人临时想到的,因为最近翻看 LAMMPS 手册时发现了一个很有意思的命令,我们先看一下文章中的高熵合金模型: 一般来说这种随机分布的高熵合金模型需要自己写代码构建,不过也听说过有一个叫 ATAT(Alloy Theoretic Automated Toolkit)的软件可以干这个事,但是我没试过。还有就是 LAMMPS 软件中有个 set 命令可以帮助我们完成这个模型构建,构建模型的 in 文件如下所示:
一般来说这种随机分布的高熵合金模型需要自己写代码构建,不过也听说过有一个叫 ATAT(Alloy Theoretic Automated Toolkit)的软件可以干这个事,但是我没试过。还有就是 LAMMPS 软件中有个 set 命令可以帮助我们完成这个模型构建,构建模型的 in 文件如下所示: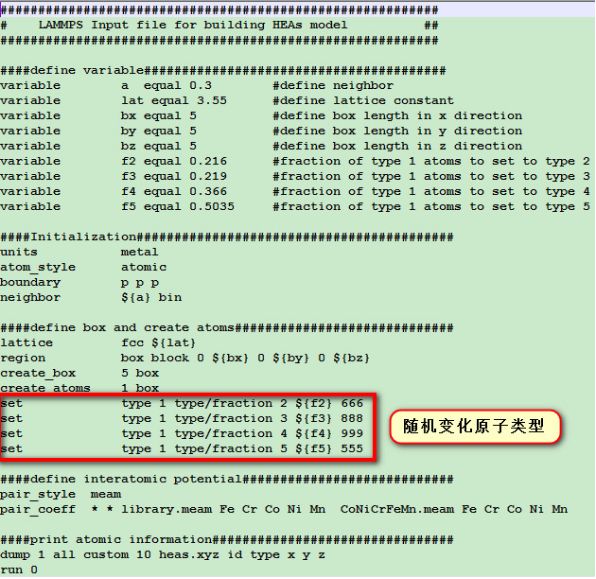 这个in文件里面所含的模型构建流程如下所示:1. 构建一定大小的盒子并按 FCC 结构填满类型为1的原子。2. 将一部分类型为 1 的原子随机替换为类型 2,3,4,5 的原子,并保证每种原子所占比例一样。3. 最后输出原子坐标信息。这里进行替换的命令即:set type 1 type/fraction 2 ${f2} 666 即在所有类型为 1 的原子中随机选取比例为f2的原子,并将这些原子的类型替换为类型 2,依次类推,直到体系中存在 5 类原子。至于 666,888 这些数都是随机设定的,只要是正整数就行。有趣的是,LAMMPS 手册中对这个命令解释时表示采用此命令进行替换,实际替换的原子数并不会完美的等于所设定的比例值,它只会接近这个值,这也是为什么我构建的体系中初始含有 500 个类型为 1 的原子,想要替换其中 100 个的原子成为类型 2 时设定的 f2 为 0.216 而不是 0.2. 最后将运行得到的文件导入 OVITO 软件中可以看到下图:
这个in文件里面所含的模型构建流程如下所示:1. 构建一定大小的盒子并按 FCC 结构填满类型为1的原子。2. 将一部分类型为 1 的原子随机替换为类型 2,3,4,5 的原子,并保证每种原子所占比例一样。3. 最后输出原子坐标信息。这里进行替换的命令即:set type 1 type/fraction 2 ${f2} 666 即在所有类型为 1 的原子中随机选取比例为f2的原子,并将这些原子的类型替换为类型 2,依次类推,直到体系中存在 5 类原子。至于 666,888 这些数都是随机设定的,只要是正整数就行。有趣的是,LAMMPS 手册中对这个命令解释时表示采用此命令进行替换,实际替换的原子数并不会完美的等于所设定的比例值,它只会接近这个值,这也是为什么我构建的体系中初始含有 500 个类型为 1 的原子,想要替换其中 100 个的原子成为类型 2 时设定的 f2 为 0.216 而不是 0.2. 最后将运行得到的文件导入 OVITO 软件中可以看到下图: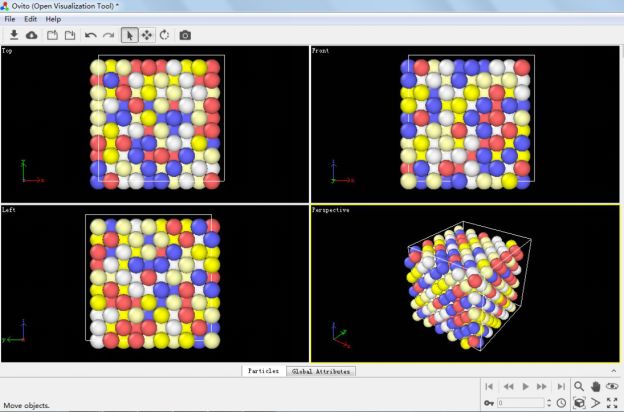 值得注意的是普通的无序混乱的高熵合金是满足不了模拟计算的要求(文章中说无序程度要符合 SRO(short-range order)参数,这个我也不是很明白~,可能要尝试构建很多种随机模型(通过修改 666,888 这些参数)后找最合适的),这里只是简单的介绍一下这个有趣的命令而已,当然这个命令用于二元合金或者是元素掺杂方面的模拟还是很有用的。●单轴拉伸过程文章中的拉伸模拟如下图所示:
值得注意的是普通的无序混乱的高熵合金是满足不了模拟计算的要求(文章中说无序程度要符合 SRO(short-range order)参数,这个我也不是很明白~,可能要尝试构建很多种随机模型(通过修改 666,888 这些参数)后找最合适的),这里只是简单的介绍一下这个有趣的命令而已,当然这个命令用于二元合金或者是元素掺杂方面的模拟还是很有用的。●单轴拉伸过程文章中的拉伸模拟如下图所示: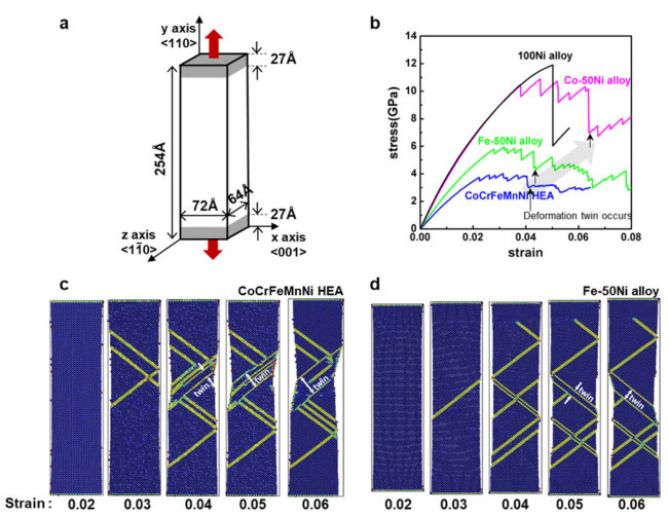 由于资源限制,这里就简单的示范一个简单的 FCC 结构金属 Ni 的拉伸过程,主要目的是介绍一下 LAMMPS 在做拉伸模拟时用到的一个核心命令以及如何得到上图所示的 Strain-Stress 关系图(b 图)以及结构变化图(c, d 图),模拟所用的 in 文件如下所示:
由于资源限制,这里就简单的示范一个简单的 FCC 结构金属 Ni 的拉伸过程,主要目的是介绍一下 LAMMPS 在做拉伸模拟时用到的一个核心命令以及如何得到上图所示的 Strain-Stress 关系图(b 图)以及结构变化图(c, d 图),模拟所用的 in 文件如下所示: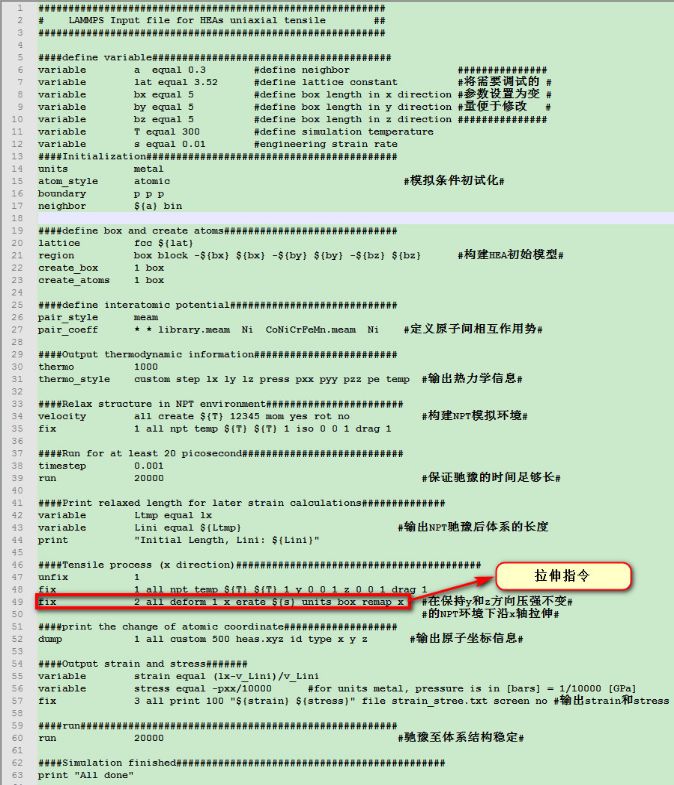 (可以考虑放大看哈~)这个 in 文件里面所含的模拟流程如下所示:1. 构建完整的 FCC 结构的金属 Ni 模型,并在 NPT 系综下达到热力学平衡状态。2. 保持不被拉伸的两个方向所受压强为 0,在 NPT 系综环境下进行拉伸操作。3. 输出原子坐标以及 Strain-Stress 信息。这里所用到最重要的命令即:fix 2 all deform 1 x erate ${s} units box remap x表示将体系沿X方向拉伸,其应变率为人为设定的参数 s。至于文章中的结构变化图,我们将得到的原子坐标文件(heas.xyz)导入至 OVITO,在 Add modification 中选取 Common neighbor analysis (俗称 CNA )即可得到模拟过程中模型结构的变化情况,如下所示:
(可以考虑放大看哈~)这个 in 文件里面所含的模拟流程如下所示:1. 构建完整的 FCC 结构的金属 Ni 模型,并在 NPT 系综下达到热力学平衡状态。2. 保持不被拉伸的两个方向所受压强为 0,在 NPT 系综环境下进行拉伸操作。3. 输出原子坐标以及 Strain-Stress 信息。这里所用到最重要的命令即:fix 2 all deform 1 x erate ${s} units box remap x表示将体系沿X方向拉伸,其应变率为人为设定的参数 s。至于文章中的结构变化图,我们将得到的原子坐标文件(heas.xyz)导入至 OVITO,在 Add modification 中选取 Common neighbor analysis (俗称 CNA )即可得到模拟过程中模型结构的变化情况,如下所示: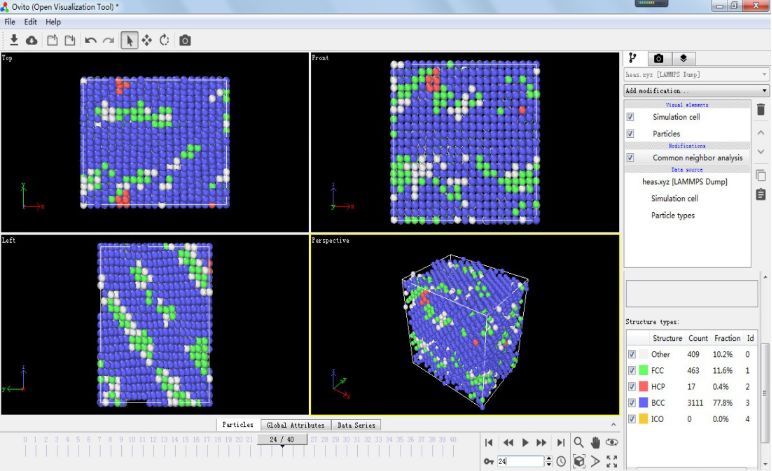 同样的,根据 LAMMPS 输出的 Strain-Stress 关系的txt文件,同样可以得到与文献中 b 图相类似的 Strain-Stress 关系图:
同样的,根据 LAMMPS 输出的 Strain-Stress 关系的txt文件,同样可以得到与文献中 b 图相类似的 Strain-Stress 关系图: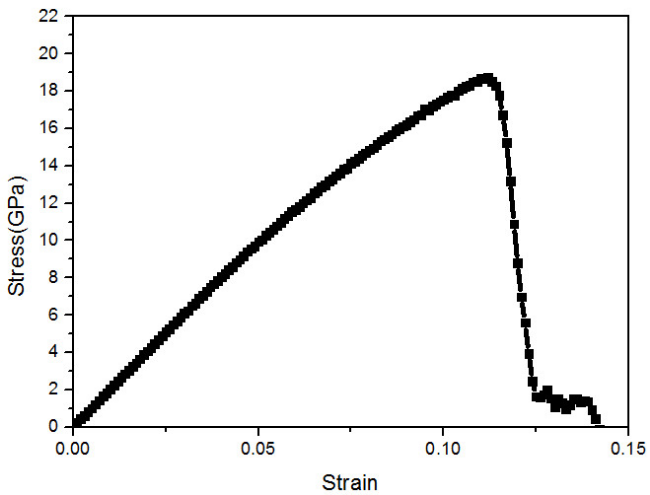 欢迎各位有兴趣的同学测试我所提供的 in 文件~此文中所有 in 文件中所涉及到的势函数文件内容均在该文献的 Supplementary Information 中可以找到。最后说一下,虽然本人不太喜欢韩国人,但是看了他们课题组的网页后,不得不承认这个研究小组的工作的确做的非常的好。在金属材料分子动力学研究领域,拟合了很多金属原子间相互作用势(拟合相互作用势通常都被认为是吃力不讨好,不愿意干的的研究工作,因为耗时长而且不容易发好文章),为金属材料的理论模拟做了很大的贡献。有兴趣的朋友可以进入他们的网站看看,他们拟合的势函数都是与 LAMMPS 软件无缝衔接的,且其网站上均有提供。他们的网站网址:http://cmse.postech.ac.kr若各位同学发现有问题的地方,欢迎留言~
欢迎各位有兴趣的同学测试我所提供的 in 文件~此文中所有 in 文件中所涉及到的势函数文件内容均在该文献的 Supplementary Information 中可以找到。最后说一下,虽然本人不太喜欢韩国人,但是看了他们课题组的网页后,不得不承认这个研究小组的工作的确做的非常的好。在金属材料分子动力学研究领域,拟合了很多金属原子间相互作用势(拟合相互作用势通常都被认为是吃力不讨好,不愿意干的的研究工作,因为耗时长而且不容易发好文章),为金属材料的理论模拟做了很大的贡献。有兴趣的朋友可以进入他们的网站看看,他们拟合的势函数都是与 LAMMPS 软件无缝衔接的,且其网站上均有提供。他们的网站网址:http://cmse.postech.ac.kr若各位同学发现有问题的地方,欢迎留言~
更多理论计算相关内容,请阅读「理论化学研习社」板块相关内容(公众号后台回复「理论」)。
加客服微信(微信号:ccl2098),申请加入研之成理理论计算交流群。


DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐

 2
2 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)