bulk转录组专题:如何获得TPM、CPM、FPKM、FPK的数据
由于gfe文件中自带.14这种的版本号,与我们转换的id不同,我们因此需要处理。此外,须获得的基因长度要与exp中的顺序大小一致。转换为以ENSEMBL(ENSG00000121410)为行名的数据框。如果你继续往下做,很可能会遇到一个报错,即使你的文件看不出任何错误。我们首先需要基因的长度。去下面网址下载以下文件。读取了参考数据,最好的方法还是将表达矩阵转为。为行名的RAW_count表达矩阵。
·
1.cpm的转换最为简单:(进行了log转换)
# exprSet是count表达矩阵
# 一句代码搞定
exprSet = log2(edgeR::cpm(exprSet)+1)2.其他形式较为复杂,这里依赖DGEobj.utils中的convertCounts函数
假设你获得了ENTREZID为行名的RAW_count表达矩阵。
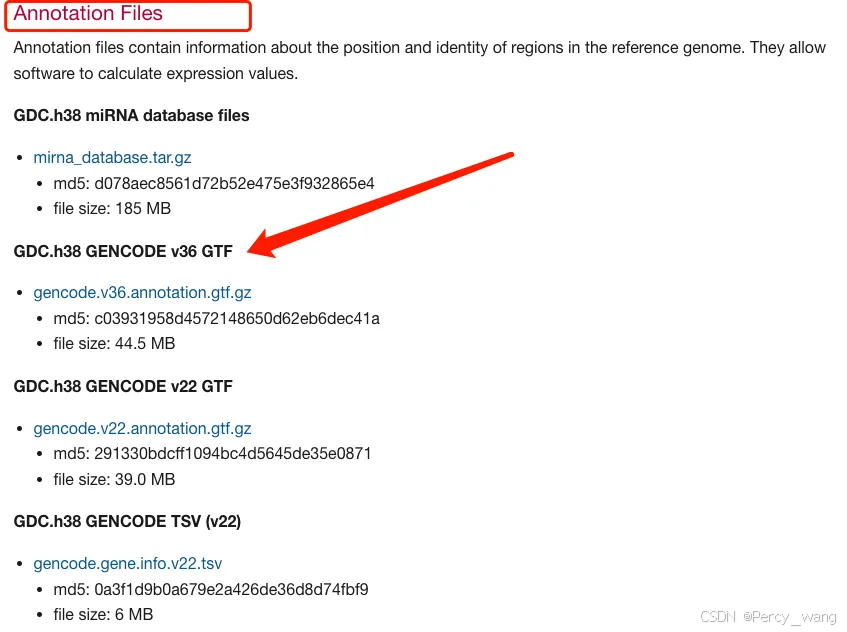
我们首先需要基因的长度。去下面网址下载以下文件。
GDC Reference Files | NCI Genomic Data Commons
2.1表达矩阵的处理
转换为以ENSEMBL(ENSG00000121410)为行名的数据框
exp=as.data.frame(exp)
exp$rownames=rownames(exp)
gene_ids=exp$rownames
# 加载 clusterProfiler 包
library(clusterProfiler)
library(org.Hs.eg.db)
# 使用 bitr 函数进行基因ID转换
converted_ids <- bitr(gene_ids,
fromType = "ENTREZID",
toType = "ENSEMBL",
OrgDb = org.Hs.eg.db)
table(table(converted_ids$ENSEMBL))
ids = distinct(converted_ids,ENSEMBL,.keep_all = T)
# 查看转换结果
exp = merge(exp,ids,by.x = "rownames",by.y = "ENTREZID")
rownames(exp)=exp$ENSEMBL
exp=exp[,-1]
exp=exp[,-45]2.2获得基因长度
if(!require(GenomicFeatures))BiocManager::install("GenomicFeatures")
library(GenomicFeatures)
txdb <- makeTxDbFromGFF("gencode.v36.annotation.gtf.gz",format="gtf")
# 获取每个基因id的外显子数据
exons.list.per.gene <- exonsBy(txdb,by="gene")
# 对于每个基因,将所有外显子减少成一组非重叠外显子,计算它们的长度(宽度)并求和
exonic.gene.sizes <- sum(width(GenomicRanges::reduce(exons.list.per.gene)))
# 得到geneid和长度数据
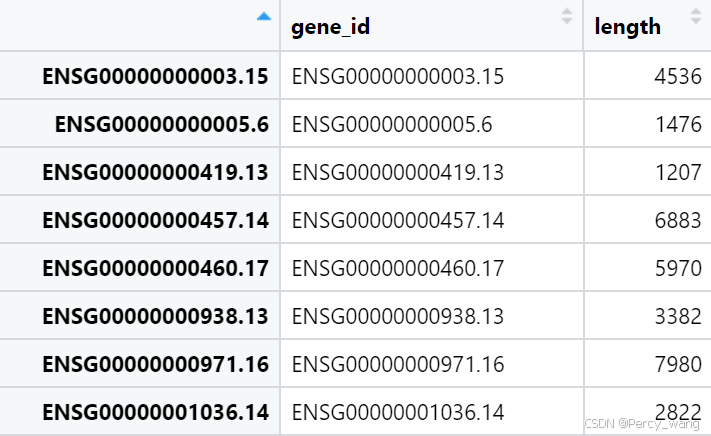
gfe <- data.frame(gene_id=names(exonic.gene.sizes),
length=exonic.gene.sizes)
head(gfe)[1:5,1:2]
# gene_id length
# ENSG00000000003.15 ENSG00000000003.15 4536
# ENSG00000000005.6 ENSG00000000005.6 1476
# ENSG00000000419.13 ENSG00000000419.13 1207
# ENSG00000000457.14 ENSG00000000457.14 6883
# ENSG00000000460.17 ENSG00000000460.17 5970
save(gfe,file = "gfe.Rdata")2.3gfe文件的处理
由于gfe文件中自带.14这种的版本号,与我们转换的id不同,我们因此需要处理。此外,须获得的基因长度要与exp中的顺序大小一致。
load("gfe.Rdata")
#提取上面处理好的基因长度列
m=rownames(exp)
gfe$gene_id2=sapply(strsplit(as.character(gfe$gene_id), "\\."), `[`, 1)
gfe[,-1]
gfe2=gfe[gfe$gene_id2 %in% rownames(exp),]
#保持顺序一致
p = identical(rownames(exp),gfe2$gene_id2);p
if(!p) {
s = intersect(rownames(exp),gfe2$gene_id2)
exp = exp[s,]
gfe2 = gfe2[s,]
}如果你继续往下做,很可能会遇到一个报错,即使你的文件看不出任何错误。

读取了参考数据,最好的方法还是将表达矩阵转为矩阵形式。
exp2=as.matrix.data.frame(exp)
effLen = gfe2$length
library(DGEobj.utils)
CC_res <- convertCounts(
exp2,
unit = "TPM", # CPM、FPKM、FPK 或 TPM
geneLength = effLen, #这里还是需要下在基因长度数据
log = FALSE, #默认为False,设为TRUE时返回Log2值
normalize = "none", #默认为none,调用edgeR的calcNormFactors()进行标准化
prior.count = NULL #为避免取0的对数,对每个观测值添加平均count。仅当 log = TRUE 时使用,
)
head(CC_res)[1:3,1:3]
CC_res[is.na(CC_res)] <- 0 #去除NA最终我们顺利得到了TPM形式:

3.写在最后。还是GEO最香啊!!

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐

 4
4 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)