PLINK-GWAS学习6------数据质控之杂合率质控
前言
杂合性:携带特定SNP的两个不同等位基因,个体的杂合率是杂合基因型的比例。个体内高水平的杂合性可能表明样本质量低(收到污染),而低水平的杂合性可能是由于近亲繁殖。
文章建议剔除杂合率偏离±3SD的个体。杂合性检查是在一组不高度相关的snp上进行的。
因此,为了生成非(高度)相关的snp列表,我们排除了高反转区域(inverse .txt [high LD regions]),并使用命令——indep- paired wise’修剪这些snp。
修剪:这是一种近似连锁平衡的标记子集的方法。在Plink中,此方法使用染色体特定窗口(区域)内SNP之间的LD强度。并根据用户指定的LD阈值,仅选择近似不相关的SNP。与聚集相比修剪操作不考虑SNP的P值。
聚集:仅识别和选择每个LD块中最重要的SNP(最低的P值)以进一步分析,这降低了剩余SNP间的相关性,同时保留了具有最强统计证据的SNP。
--indep
--indep-pairwise
--indep-pairphase
这些命令产生一个修剪过的SNP子集,这些标记之间大致处于连锁平衡状态,这些SNP的id被写入plink.prune.in,并且所有被排除的SNP的id被写入plink.prune.out。他们目前基于基因型等位基因数的相关性所产生,未考虑位置(phase)。(结果可能与plnik1.07版本有稍微不同,由于存在缺失数据时r2计算的一个小bug,以及对多重共线性的更系统的处理。)在那后续的PLINK运行时,输出文件是一个有效的–extract/–exclude命令的输入文件。--indep需要三个参数,以变体数或者千碱基数(当出现“kb”字符时)为单位的窗口大小,在每一步结束时窗口需要移动的变体数,和一个方差膨胀因子阈值(VIF)。在每一步中,当前窗口中所有VIF超过阈值的突变体都会被移除。--indep-pairwise的前两个参数与 --indep相同。它的第三个参数是一个成对的r2阈值:在每一步中,在当前窗口中平方相关(squared correlation)超过阈值的突变体对将被标记,并且突变体会从窗口中修剪掉直到没有这样的突变体对存在。
最后--indep-pairphase与--indep-pairwise几乎相同,除了他的r2值是基于最大似然估计得出的(like “–r2 dprime” below)--extract通常接受一个有突变体ID的文本文件(通常一个一行,但是用空格隔开也没关系),并且去除所有不在列表中的突变体。使用’range’修饰符,输入文件应该改为set range格式。--exclude对所有列出的变体执行相同的操作。注意,当主输入文件集包含重复的变体id时,这与PLINK 1.07的行为略有不同:PLINK 1.9删除所有匹配,而PLINK 1.07只删除一个匹配的变体。如果您的意图是解析重复,您现在应该使用——bmerge而不是——exclude。
plink --bfile HapMap_3_r3_9 --exclude inversion.txt --range --indep-pairwise 50 5 0.2 --out indepSNP
inversion.txt
第一列染色体号,第二列起始位置,第三列终止位置,第四列set ID,第五列组标签


plink --bfile ../04.step4/HapMap_3_r3_9 --extract indepSNP.prune.in --het --out R_check


杂合率分布图
check_heterozygosity_rate.R
het <- read.table("R_check.het", head=TRUE)
pdf("heterozygosity.pdf")
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
hist(het$HET_RATE, xlab="Heterozygosity Rate", ylab="Frequency", main= "Heterozygosity Rate")
dev.off()
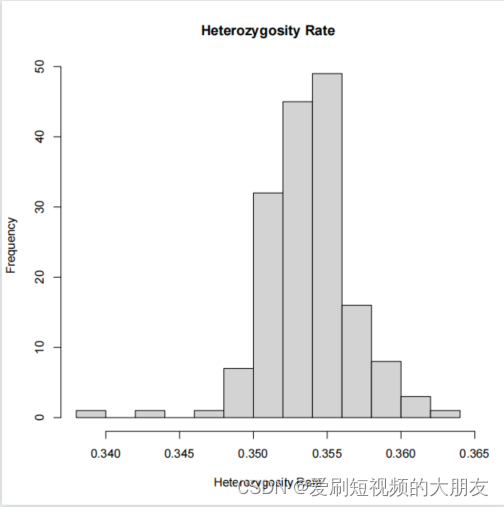
下面的代码生成了一个与杂合度平均偏差超过3个标准差的个体列表。
het <- read.table("R_check.het", head=TRUE)
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
het_fail = subset(het, (het$HET_RATE < mean(het$HET_RATE)-3*sd(het$HET_RATE)) | (het$HET_RATE > mean(het$HET_RATE)+3*sd(het$HET_RATE)));
het_fail$HET_DST = (het_fail$HET_RATE-mean(het$HET_RATE))/sd(het$HET_RATE);
write.table(het_fail, "fail-het-qc.txt", row.names=FALSE)
.#当使用我们的示例数据/ HapMap数据时,这个列表包含2个个体(即,两个个体的杂合度与平均值的偏差超过3sd)。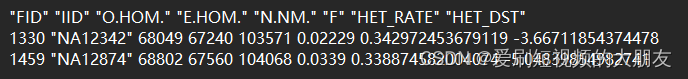
修改此文件,使其与PLINK兼容,删除文件中所有引号,只选择前两列。
sed 's/"// g' fail-het-qc.txt | awk '{print$1, $2}'> het_fail_ind.txt

#去除杂合度异常值
plink --bfile HapMap_3_r3_9 --remove het_fail_ind.txt --make-bed --out HapMap_3_r3_10

这是关于het文件格式的描述 第三列是观测杂合数,第四列是期望杂合数,第五列是观察到的非缺失、非单型的SNP数量,第六列是估计的F值
第三列是观测杂合数,第四列是期望杂合数,第五列是观察到的非缺失、非单型的SNP数量,第六列是估计的F值
大佬贴出了F值得计算方法如下:(原贴请搜CSDN:笔记 GWAS 操作流程2-5:杂合率检验)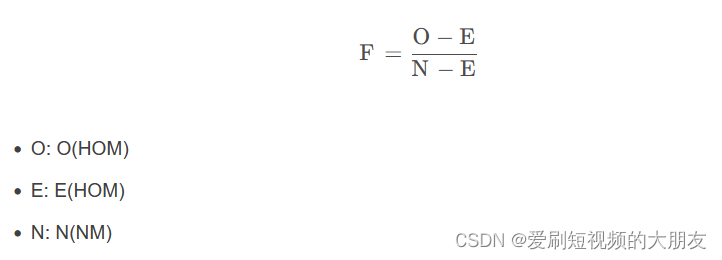

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐


 0
0 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)