iMetaOmics | 基因组所刘永鑫组-易扩增子(EasyAmplicon):用户友好的扩增子测序数据分析指南...
点击蓝字 关注我们易扩增子(EasyAmplicon):用户友好的扩增子测序数据分析指南iMeta主页:http://www.imeta.science方法论文●原文链接DOI: https://doi.org/10.1002/imo2.42●2024年11月20日,中国农业科学院深圳农业基因组研究所刘永鑫团队在iMetaOmics在线发表了题为“Unveiling microbial com..
点击蓝字 关注我们
易扩增子(EasyAmplicon):用户友好的扩增子测序数据分析指南

iMeta主页:http://www.imeta.science
方法论文
● 原文链接DOI: https://doi.org/10.1002/imo2.42
● 2024年11月20日,中国农业科学院深圳农业基因组研究所刘永鑫团队在iMetaOmics在线发表了题为“Unveiling microbial communities with EasyAmplicon: A user-centric guide to perform amplicon sequencing data analysis”的文章。
● 本研究方案论文为易扩增子(EasyAmplicon)的实验说明手册,对EasyAmplicon分析流程中每一步骤做了详尽的解释说明及操作演示,为新手使用者提供了参考。
● 第一作者:Salsabeel Yousuf、罗豪、曾美尹
● 通讯作者:刘永鑫(liuyongxin@caas.cn)
● 主要单位:中国农业科学院深圳农业基因组研究所
亮 点
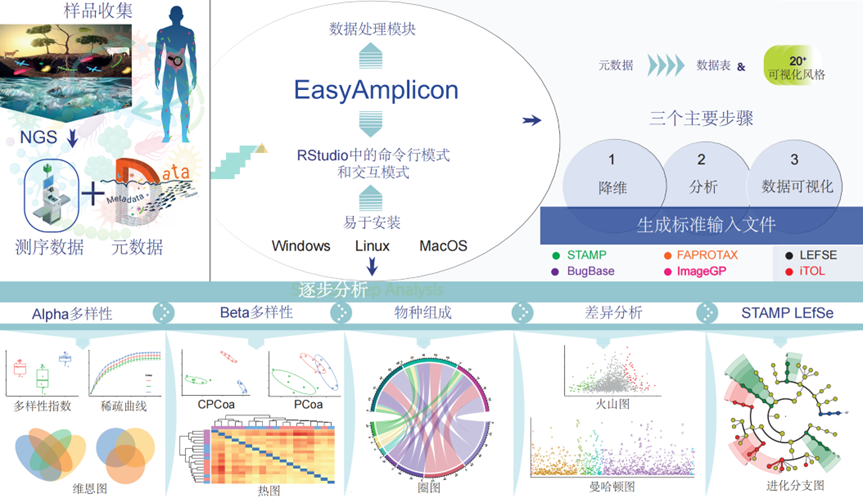
● EasyAmplicon是扩增子测序的一个全面、自动、用户友好的分析流程,无缝集成了各种生物信息学工具;
● 该流程支持高效的数据预处理、多样性分析和高级可视化操作,使更多的研究人员得以进行高通量数据分析;
● 每个步骤都配有说明、故障排除提示和解决方案,以方便用户使用。
摘 要
二代测序(NGS)的出现彻底改变了微生物组研究,人们能够通过扩增子测序深入探索微生物群落。成本的下降加上测序在不同领域的广泛应用,加剧了人们对经过验证的、自动化的、可重复的和适应性强的分析流程的迫切需求。然而,分析这些高通量数据往往需要大量的生物信息学知识,这给许多研究人员造成了阻碍。针对这一点,我们在2023年开发了EasyAmplicon,这是一款全面的、用户友好的流程,集成了USEARCH和VSEARCH等流行工具,提供从原始数据到结果的简化工作流程。值得注意的是,EasyAmplicon在推出一年内获得了广泛的认可,迄今为止已被引用127次。为了进一步方便研究人员使用,我们提供了EasyAmplicon的详细使用手册和视频教程,指导用户完成该流程的每个步骤,包括数据预处理(质量过滤、嵌合体去除)、扩增子序列变体(ASV)分析、多样性分析和数据可视化。该流程易于使用,每个步骤都有文档记录,使研究人员无需复杂的生信技能即可执行。该EasyAmplicon流程可在GitHub上免费获取(https://github.com/YongxinLiu/EasyAmplicon)。
视频解读
Bilibili:https://www.bilibili.com/video/BV1zqqTYHEwx/
Youtube:https://youtu.be/p6iovVInAUI
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/imetaomics/
全文解读
引 言
二代测序(NGS)技术被广泛应用于各种环境中的微生物群落研究,例如人类微生物组计划(https://www.hmpdacc.org/hmp/index.php)和地球微生物组计划(https://earthmicrobiome.org/)等。在过去的二十年里,NGS技术、数据分析方法和生物信息学工具的进步在我们对人类、动物、植物和环境微生物组的研究中,以及它们之间相互作用的理解中发挥了关键作用。这些进展还应用于环境微生物监测,以及揭示微生物组与人类疾病之间的关联。其他应用包括分析细菌基因组序列以识别抗菌素耐药性基因和评估细菌丰度以识别致病的潜在菌群失调。
在过去十年中,扩增子16S核糖体RNA(16S rRNA)和宏基因组分析的框架已经很好地建立起来。16s rDNA基因亚基测序是一种公认的表征微生物群落多样性的方法,并且为细菌、古细菌和一些真核生物的全基因组测序提供了一种经济有效的替代方法。扩增子测序已成为研究微生物群落不可缺少的工具,使研究人员能够分析各种环境下微生物组的组成和功能。流行的扩增子分析管道,如Mothur、USEARCH和QIIME,提供了强大的质量控制、序列处理、操作分类单元(OTU)聚类、多样性分析和分类学分类功能,显著推进了微生物组研究。尽管取得了这些进展,但生物信息学的复杂性和对广泛生物信息学知识的需求仍然是许多研究人员的障碍。
为了应对这些挑战,去年我们推出了EasyAmplicon,这是一种用户友好且全面的扩增子分析流程,旨在简化分析过程的同时保持强大的功能。EasyAmplicon将多种流行工具集成到一个简化的工作流程中,指导用户从原始数据获取结果。通过降低技术壁垒,该流程旨在使生物信息学背景有限的研究人员也能进行高级扩增子分析,从而拓宽微生物群落研究的范围和影响。EasyAmplicon包含用于高效数据处理和可视化的模块,它生成与流行软件包例如STAMP、LEfSe、PICRUSt 1 & 2、BugBase、FAPROTAX、ImageGP和iTOL兼容的标准输入文件。在这里,我们展示了整个流程,详细介绍了从安装和使用到最终可视化阶段的每个步骤,并指定了所需的输入文件和输出生成的数据。
应用
EasyAmplicon已成功应用于各种研究项目,展示了其多功能性和实用性。值得注意的应用包括土壤微生物组,研究人员已使用EasyAmplicon来分析农业土壤中的微生物多样性,揭示了土壤健康与营养循环之间的联系。在肠道微生物群研究中,该流程促进了对人类和动物肠道微生物群的研究,有助于了解健康和疾病之间的关系。EasyAmplicon还被用于探索淡水和海洋环境中的微生物多样性,为生态和环境研究做出了贡献。这些例子凸显了EasyAmplicon在不同生态系统微生物群落研究中的广泛适用性。
局限性
虽然EasyAmplicon主要用于分析第二代测序平台(例如Illumina,Ion Torrent)生成的16s rRNA扩增子测序数据,但它与某些第三代测序(TGS)数据(例如PacBio)的兼容性有限。TGS的功能可能会受到限制,但未来的更新旨在解决这一问题,以确保与TGS数据完全兼容。
生物信息学流程描述和方法
EasyAmplicon流程和图形用户界面(GUI)web服务器ImageGP(https://www.bic.ac.cn/ImageGP/)通过QIIME,USEARCH,VSEARCH和内部脚本进行组合。这些工具的合作可以评估序列质量,拆分条形码序列,去除测序背景,并识别ASV和OTU。
设备要求
● 操作系统要求:64位Linux(例如,Ubuntu 16.04+或CentOS 7.5+)。
● 硬件要求:至少4 GB的RAM和30 GB的可用磁盘空间(图 1A)。
扩增子数据
用户可以利用在线存储库中的现有数据来进行微生物组分析,也可以通过将样本(例如动物、植物、人类或土壤)交到测序公司来生成自己的数据(图 1B)。
软件和数据库
视频教程链接:https://youtu.be/ZFrqi5P2i8MTo
1. 根据系统(Windows/Mac/Linux)安装依赖项软件,如图1C所示。
2. Git for Windows 2.x. x:该工具为Windows用户提供了运行shell命令的环境,可以在RStudio Terminal(http://gitforwindows.org/)中使用。
3. RStudio 2023.xx:R的集成开发环境。选择适合您系统的版本(Windows 10+或 macOS 10.14+)并下载最新版本(https://posit.co/download/rstudio-desktop/)。
4. 体积小、启动速度快的纯文本编辑器(https://www.editplus.com/)。
5. R:用于数据分析和可视化的统计编程语言(https://www.r-project.org/)。Windows用户可能需要Rtools安装R包(https://cran.rproject.org/bin/windows/Rtools/)。
6. STAMP v2.1.3或更高版本:该软件用于分析微生物分类和功能谱(http://kiwi.cs.dal.ca/Software/STAMP)。
7. QIIME 2:可扩展的、免费的、开源的二代微生物组生物信息学平台(https://qiime2.org)。
8. Conda:软件管理系统(https://repo.anaconda.com/miniconda)。
9. USEARCH v10.0.240或更高版本:USEARCH是一款多功能高通量序列分析工具,提供聚类、嵌合体检测和OTU筛选等功能。它被广泛用于微生物组研究,可高效处理大型数据集(http://www.drive5.com/usearch)。
10. VSEARCH v2.7.1+52或更高版本:VSEARCH是一款开源工具,用于执行序列聚类、嵌合体检测和去重等任务。它是USEARCH的流行替代品(https://github.com/torognes/vsearch)。
注意:用户既可以通过官网链接依次下载软件,也可以直接从百度网盘(https://pan.baidu.com/s/1Ikd_47HHODOqC3Rcx6eJ6Q?pwd=0315)选择下载。
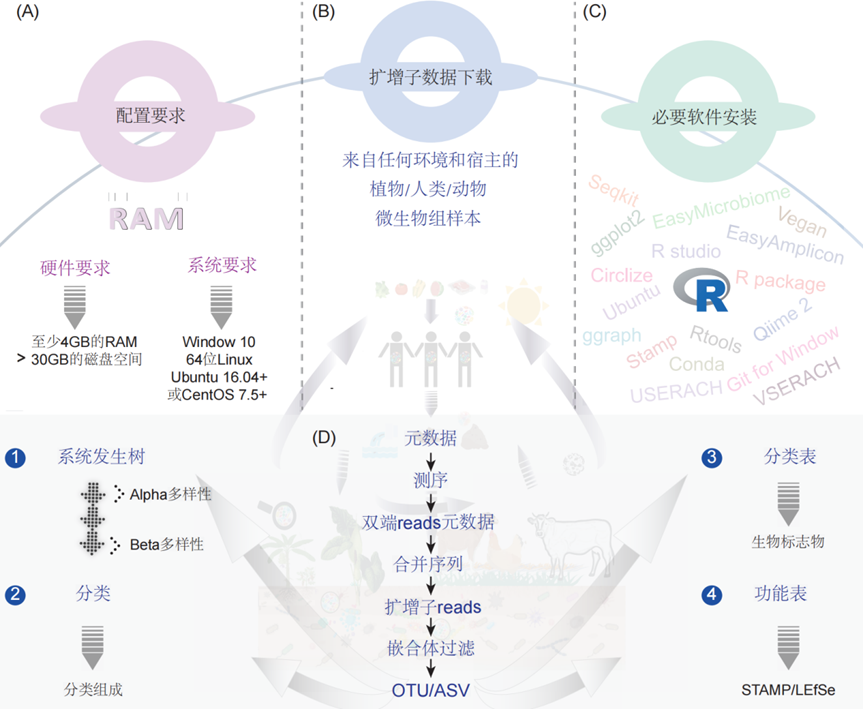
图1. EasyAmplicon流程的系统要求和一般工作流程
(A)要运行此流程,系统必须满足以下要求:至少4GB的RAM和30GB以上的可用磁盘空间。操作系统:Windows 10(64位)或兼容的Linux发行版(特定的Ubuntu或CentOS版本)。(B)EasyAmplicon流程与各种来源的测序数据兼容,包括动物、植物、人类和土壤样本。(C)下载/安装软件和数据库,如GitBash、R studio、R tools、Ubuntu、VSEARCH、USERACH、STAMP和Qiime2。(D)该流程的整体工作流程主要包括三个步骤。(i)降维是初始步骤,其处理原始测序reads并将其转换为特征表。特征表总结了测序样本中每个样本发现的生物特征的丰富程度(例如,操作分类单元OTU或扩增子序列变体ASV)。(ii)分析过程中,在上一步中生成的特征表将作为各种下游分析的基础。系统发育分析探索样本中鉴定的生物之间的进化关系。分类步骤将分类标签(例如种或属)分配给特征表中的OTU或ASV。功能预测分析利用遗传数据预测样本中存在的微生物的潜在功能作用。Alpha多样性量化单个样本内的物种多样性,而Beta多样性衡量样本间的物种多样性。(iii)统计和可视化是最后一步,侧重于生成适合发表的高质量图表,进行分析结果的可视化。并采用统计检验来解释研究结果的生物学意义。该流程生成各种输出图表以供进一步生物学解释,包括描述进化关系的系统发育树、详细描述微生物群落组成的分类学概况、预测潜在代谢功能的功能概况,以及分别量化样本内和样本间物种多样性的Alpha和Beta多样性指标。重要的是,我们的流程生成的输入文件与其他流行的微生物组分析工具(如STAMP和LEfSe以及FAPROTAX)兼容,可供进一步探索。
EasyAmplicon安装
要使用EasyAmplicon进行扩增子数据分析,您需要下载EasyAmplicon软件及其附带的数据库EasyMicrobiome。我们强烈建议用户从官方GitHub网站https://github.com/YongxinLiu/EasyAmplicon下载。这可确保用户拥有最新、最安全版本的软件和数据库用于扩增子分析。从GitHub下载是一个简单的过程:只需访问EasyAmplicon/EasyMicrobiome存储库,点击“Code”按钮,然后选择“download ZIP”将下载的文件保存到本地磁盘(例如C盘或D盘)上即可。确保下载后目录名保持不变;对于EasyAmplicon,保持目录名称为“EasyAmplicon‐master”。对于数据库目录,解压缩文件后,将解压缩的文件夹从“EasyMicrobiome‐master”重命名为“EasyMicrobiome”。另外也可通过百度网盘https://pan.baidu.com/s/1Ikd_47HHODOqC3Rcx6eJ6Q?pwd=0315(必须先创建一个帐户)一键下载EasyAmplicon和相关软件。
注意:因为软件链接经常更新,建议从GitHub获取最新的BaiduNetdisk链接。
下载R包
在EasyAmplicon统计和可视化的过程中可能需要用到500多个R包(https://cran.r-project.org/web/packages/available_packages_by_name.html#available-packages-B)。为简化安装过程,用户可以直接从此链接下载所需的R包:http://www.imeta.science/db/win/4.3.zip。或者,用户也可以从百度网盘(https://pan.baidu.com/s/1Ikd_47HHODOqC3Rcx6eJ6Q?pwd=0315)下载R包,位于db/win/4.x.zip或db/mac/R4.x_mac_libraryX86_64.zip中。随后解压并将其放在R的library目录下即可完成安装。4.x.zip包为常用R包的集合,使用户可以绕过R包的逐个下载和安装。要想知道本机R的library路径,可在启动RStudio后,在菜单“Tools”中通过“install packages”选项查看该版本R的R包默认安装目录。将压缩文件4.x.zip复制到win-library目录。选择4.x.zip,右键单击,然后选择“解压到当前文件夹”。如果提示替换文件,请选择“全部替换”。替换后重新启动RStudio,并在“>”在左下角的控制台中输入“library (ggplot2)”进行测试,如果出现下面的信息,则表示安装成功。
[Workspace loaded from /var/www/html/ehbio_doc/train/ .RData]
> library (ggplot2)
如果出现类似“Error in library (‘ggplot2’): there is no package called ‘ggplot2’”的错误,则表示R包未成功安装。这可能是由于解压不成功或文件夹放置错误。请仔细检查并重复上述步骤,或者也可以使用“install.packages”命令安装新包,例如:
install.packages("devtools ")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("edgeR")
library(devtools)
devtools::install_github("microbiota/amplicon")
对于Mac用户,请从“Mac”目录中提取压缩文件夹到下载文件夹。然后,执行如下命令:
cp -r ~/Downloads/library/*
/Library/Frameworks/R.framework/Versions/4.x/Resources/library/
注意:如果在工作目录(EasyAmplicon)、数据库目录(EasyMicrobiome)或4.3 packages文件夹中存在子文件夹,可能会导致EasyAmplicon在代码执行期间遇到错误。这些位置中的嵌套目录结构可能会破坏文件路径,阻碍脚本调用必要文件。
扩展软件和默认数据库
运行本流程所需的各种软件工具和数据库均配置在数据库目录EasyMicrobiome中,用户可直接下载使用,也可从官方网站(包括GitHub、百度网盘)获取最新版本EasyMicrobiome。本流程用到了很多软件工具和数据库,例如UNITE、RDP、SILVA、GreenGenes等,这些数据库为细菌序列的分类和鉴定提供了重要的信息。
UNITE数据库(https://unite.ut.ee/)用于真菌或真核生物的内部转录间隔区(ITS)测序分析,默认保存在EasyMicrobiome/usearch目录中。核糖体数据库项目(RDP)默认位于EasyMicrobiome数据库文件夹中。用户可以按照QIIME 2分析部分提供的说明下载并安装SILVA 和GreenGenes数据库以进行16S微生物组分析。或者,用户也可以从USEARCH的官方网站(http://www.drive5.com/usearch/manual/sintax_downloads.html)下载与其兼容的GreenGenes数据库(https://ftp.microbio.me/greengenes_release/current/)。SILVA数据库也可以以多种格式下载,官网https://www.arb-silva.de/download/提供了说明和下载选项。
输入数据文件
本流程的输入数据文件包括实验设计元数据和原始测序数据。本文中使用的原始测序数据以压缩的FASTQ格式保存在“seq”文件夹中,可通过元数据从基因组序列档案库(GSA)下载备份,网址为https://ngdc.cncb.ac.cn/gsa/。虽然本流程假定原始测序数据存储在“seq”文件夹中并以压缩的FASTQ格式提供,但是,根据特定的数据存储和格式要求,也可能需要进行调整。如果用户更喜欢使用Web服务器,建议将数据存放在GSA中,因为这可以大大减少下载时间。或者,也可以使用公共数据中心,例如美国国家生物技术信息中心(NCBI)和欧洲生物信息学研究所( https://www.ebi.ac.uk/),它们也可以提供在线分析的下载链接。
wget -c ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz -O seq/KO1_1.fq.gz
gunzip *.gz
Conda的下载与安装
EasyAmplicon脚本命令的执行是在Git Bash终端内进行的,而各种分析命令是在Linux下的Windows子系统(Windows Subsystem for Linux,WSL)的Bash终端内执行的。要使用WSL,用户需要执行一系列步骤。首先,在计算机“设置”中键入“启用或关闭Windows功能”,然后启用“适用于Linux的Windows子系统”。接下来,在Microsoft应用商店中搜索Linux发行版(例如Ubuntu),然后进行安装。安装成功后,在Ubuntu WSL环境中打开终端并创建帐户。成功激活Ubuntu后,开始安装并激活Conda以获取Conda环境。
(i)为了下载并安装Conda,打开Ubuntu并运行以下命令:
wget -c https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh -b -f
~/miniconda3/condabin/conda init
(ii)初始化Conda:关闭并重启终端窗口。然后,继续安装QIIME 2。此方法利用Conda管理器在线安装QIIME 2。首先定义所需的QIIME版本;这里,我们将使用qiime2-amplicon-2024.5作为示例。
n=qiime2-amplicon-2024.5
接下来,下载QIIME安装过程所需的软件依赖列表:
wget -c https://data.qiime2.org/distro/amplicon/${n}-py39-linux-conda.yml
如果主源发生故障,请运行以下命令作为替代:
wget -c http://www.imeta.science/db/conda/${n}-py39-linux-conda.yml
随后,要创建一个以所选QIIME版本命名的新Conda环境并安装下载的YAML文件中列出的依赖项:
conda env create -n ${n} --file ${n}-py39-linux-conda.yml
下一步骤(约1.2 GB)是可选操作,目的是打包已安装的环境以方便用户将其上传到计算机服务器上。
conda pack -n ${n} -o ${n}.tar.gz
最后,激活并初始化QIIME 2环境:
conda activate ${n}
注意:如果用户在此步骤中遇到困难,可以打开Web浏览器并访问Miniconda网站(https://docs.conda.io/en/latest/miniconda.html),该网站提供了全面的安装说明。找到与您的操作系统(例如Windows、macOS、Linux)相对应的部分,在该部分中,搜索下载最新的Miniconda3安装包。文件名通常包含“Miniconda3-latest”和用户系统类型(例如“x86_64”)。如有必要,请将Linux-x86_64替换为您的特定类型(例如,macOS的osx-x86_64)。用户可以在Miniconda下载页面上根据系统配置找到目标下载链接。对于Windows 10 plus上的Ubuntu用户,请下载Linux 64位安装程序。
R设置
● 以Windows 10+为例
● 打开Rstudio主界面,选择Tools——Global Options选项
● 设置General——Default working directory(设置默认工作目录)
● 点击Browse选择C:/amplicon(即您要工作的文件夹的位置,可以任意命名)
● 选择编码格式:解决中文乱码问题
● Code——Saving——Default text encoding——选择UTF-8
● 选择镜像:加快安装包下载(可选步骤)
● Packages——Primary CRAN repository——CRAN mirror——选择Beijing,或者合肥/广州/兰州/上海(选择离您所在地最近的CRAN镜像)
● 同样按此路径切换设置,设置终端为Git Bash
● 打开RStudio——Tools——Global Options——Terminal——New Terminal——Git Bash——OK)。关闭文件,重启Rstudio。
● 在R studio设置工作目录:
● 选择Session菜单——Set Working Directory——Choose Directory打开工作文件夹所在的目录
● 选择File菜单——Open File——进入EasyAmplicon文件夹——打开pipeline.sh(windows/linux)或者pipeline_mac.sh(mac),备有中英文版本,选择合适个人的即可
总结
EasyAmplicon允许灵活的数据输入,接受各种格式,如双端或单端测序reads(FASTQ)、干净扩增子(FASTA)。流程通常从合并双端reads、移除引物和Barcode以及过滤低质量reads以获得干净扩增子开始,主要使用VSEARCH或USEARCH软件。然后,干净的扩增子可以基于97%的相似性聚类到OTU中,使用从头聚类的方法去噪为ASV,或直接映射到GreenGenes等参考数据库以获取有参聚类OTU表。OTU/ASV生成后,EasyAmplicon会构建一个特征表,总结每个样品中生物特征的丰度。该特征表与由代表性序列构建的系统发育树相结合,可推动下游分析,例如Alpha和Beta多样性分析、物种注释、微生物功能预测和差异特征发现,以突出样本组之间的区别性特征(图 1D)。为了更好地理解,用户可以参考每个部分提供的视频教程。
教程
视频链接教程:https://www.youtube.com/watch?v=9ORamd84hUc。
开始
本节详细介绍了EasyAmplicon工作环境准备的步骤。首先将工作目录(wd)设置为amplicon/EasyAmplicon,将软件数据库目录(db)设置为EasyMicrobiome。请勿直接使用从GitHub下载的文件作为工作目录。相反,请创建一个名为 amplicon/EasyAmplicon的空文件夹并将其指定为工作目录。按照脚本(中文脚本为pipeline.sh、英文为pipe_en.sh、Mac用户为pipe_mac.sh)中提供的说明正确设置$wd和$db的路径。在RStudio中逐行或逐段执行代码以完成分析过程。注意每次打开RStudio时,都需要重新运行以下四行代码来建立工作目录和数据库目录。
wd=/c/EasyAmplicon
db=/c/EasyMicrobiome
PATH=$PATH:${db}/win
cd ${wd}
注意:用户也可以将$wd和$db替换为EasyAmplicon和EasyMicrobiome的安装路径,C盘和D盘均可。有关设置环境变量的详细说明可参考脚本获取。
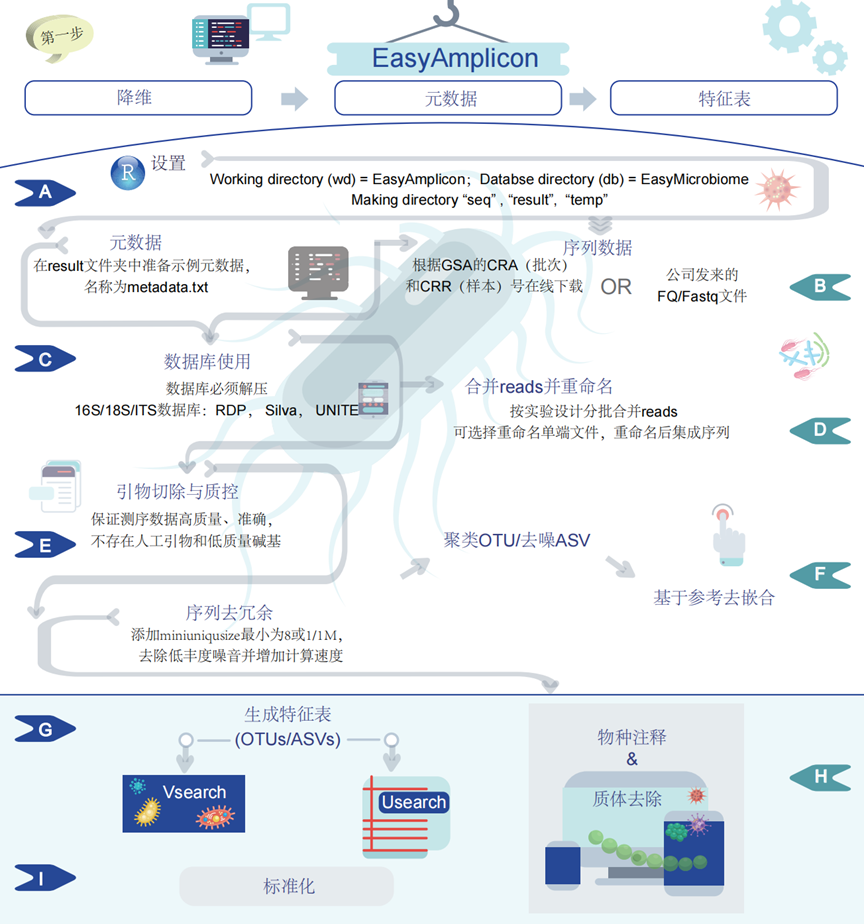
图2. 在Pipeline中,第一步是降维,将原始数据处理成特征表
(A)首先,为EasyAmplicon设置工作目录(wd)并为EasyMicrobiome设置数据库目录(db)。(B)用户必须将“metadata.txt”保存在“result”目录中,并将测序数据FASTQ文件保存在“seq”目录中,通常以.fq.gz结尾,每个样本一对文件。如果需要,使用gunzip解压缩.gz文件(VSEARCH通常可以直接读取它们)。(C)EasyAmplicon利用预先构建的参考数据库进行序列分析。这些数据库包括RDP、SILVA和UNITE(默认情况下,保存在EasyMicrobiome文件夹中)。第一次使用数据库时必须解压,您可以在往后跳过此部分。(D)然后,根据实验设计合并和重命名reads。(E)随后,此步骤涉及严格的质量控制措施,根据各种标准(例如测序错误和碱基质量得分)过滤掉低质量reads。(F)执行序列去重复以删除重复序列,并进行聚类或去噪以将相似序列分别分组为操作分类单元(OTU)或扩增子序列变体(ASV)。此外,基于参考的嵌合体检测有助于识别和消除伪影。(G)使用USEARCH和VSEARCH等软件工具生成特征表,其中包含样本中微生物特征的丰度信息。(H)根据参考数据库(如RDP、SILVA 或UNITE)应用分类注释,同时排除非目标DNA序列(质体)。随后,过滤表格以删除低质量序列。(I)接下来进行标准化以进行比较和分析。这种全面的流程能够高效处理和分析扩增子测序数据,从而准确表征微生物群落特征。
降维(从原始数据到表格)
准备文件
运行该流程需要两个主要数据文件进行分析,包括:测序数据和元数据文件。测序数据(*.fq.gz)是seq/目录中的成对FASTQ文件,通常以.gz压缩格式保存以节省空间。元数据(metadata.txt)是与样本编号相对应的分组、时间、位置等描述信息。打开pipeline.sh,它默认保存在从Github或百度下载的EasyAmplicon master文件夹中。然后通过运行此代码在工作目录中创建子目录,用于测序数据储存(“seq”)、结果储存(“result”)和临时储存(“temp”)(图 2A)。
mkdir -p seq result temp
注意:建议将测序数据保存在seq目录中,将元数据原始文本保存在result目录中。
元数据/实验设计
本节指导用户检查和准备样本元数据文件以供下游分析。
(1)验证元数据结构
“csvtk”包检查元数据文件的结构和格式(图 2B),以确定表中的行数(即样本数)和列数。在RStudio中运行csvtk -t stat result/metadata_raw.txt。故障提示:如果用户在运行代码时遇到错误“CSVTK not found”,则表示您的系统上没有csvtk命令行工具。默认情况下,csvtk保存在数据库目录内的EasyMicrobiome/win文件夹中。确保主文件夹中没有嵌套的EasyMicrobiome子文件夹,因为这可能会破坏文件路径,阻碍脚本调用必要文件。
(2)检查文件格式
要查看文件头,请使用下面提供的cat命令。head命令用于显示文件头,“-n3”选项将输出限制为元数据文件的前3行,以便您验证列标题及样本名。
cat -A result/metadata_raw.txt | head -n3
注意:如果您是Windows用户,您可能会注意到数据末尾有一个“^M”符号,它代表Windows特有的回车符。下一步会对其进行格式化。
(3)格式化元数据文件(Windows专用)
此步骤解决了特定于Windows系统的潜在格式问题。“sed”命令用于删除在Windows系统上创建的元数据文件中可能存在的回车符(\r)。虽然这些字符在查看文件时通常不可见,但可能会在分析过程中引起问题。运行以下代码,将result/metadata_raw.txt替换为result/metadata.txt,后者应为包含样本信息的制表符分隔文本文件。
sed 's/\r//' result/metadata_raw.txt > result/metadata.txt
接下来,运行下面给出的代码,再次使用cat命令查看清理文件的前几行并确认回车符是否删除。
cat -A result/metadata.txt | head -n3
通过这些步骤,元数据文件将被正确格式化并可供下游分析使用。
测序数据
本节介绍使用EasyAmplicon分析所需的测序数据的两种方法。
(1)从公共资源下载
如果测序数据不易获得,EasyAmplicon允许用户从公共存储库(如SRA)下载。以下是使用wget命令下载特定文件的示例:
wget -c ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz -O seq/KO1_1.fq.gz
“awk”命令可以根据实验设计编号进行批量下载和重命名。下面给出的命令用于为每个样本构建下载URL。
awk '{system("wget -c ftp://download.big.ac.cn/gsa/"$5"/"$6"/"$6"_f1.fq.gz -O seq/"$1"_1.fq.gz")}' \
<(tail -n+2 result/metadata.txt)
awk '{system("wget -c ftp://download.big.ac.cn/gsa/"$5"/"$6"/"$6"_r2.fq.gz -O seq/"$1"_2.fq.gz")}' \
<(tail -n+2 result/metadata.txt)
注意:公共测序数据存储库提供各种数据集。请参阅特定存储库文档以了解下载说明和数据格式。
(2)利用现有的测序数据
此步骤介绍如何下载和准备测序数据以在RStudio中进行分析。如果已有测序数据,请确保将FASTQ文件(以.gz扩展名压缩)(例如KO1_1.fq.gz)放在工作目录EasyAmplicon内的seq目录中。注意确保文件名与元数据文件中的样本ID相对应,以便进行准确分析(图 2B):
ls -sh seq/
zless seq/KO1_1.fq.gz | head -n4
请运行此代码可查看每行前60个字符:
zless seq/KO1_1.fq | head | cut -c 1-60
提供的命令如“ls -sh seq”和“zless”有助于管理和查看seq目录中的压缩文件。要确定有关测序数据的基本统计信息(例如读取次数及其长度),请运行以下代码,该代码依赖seqkit程序:
seqkit stat seq/KO1_1.fq.gz
seqkit stat seq/*.fq.gz > result/seqkit.txt
head result/seqkit.txt
数据库
这一部分重点介绍EasyAmplicon流程中下游分析所使用的参考数据库。它们的位置取决于您如何下载它们。如果您通过百度网盘下载了EasyAmplicon数据库,它们可能存储在名为“db”的目录中。相反,如果您从GitHub下载它们作为EasyMicrobiome的一部分,则可以在“win”文件夹中找到它们。文件夹通常包含常用的参考数据库;但是,某些数据库(如EzBioCloud)可能未包含在内。如果您需要特定数据库,您可以自行从百度网盘(如果可用)或可信的在线存储库下载。下载后,将数据库文件放在EasyMicrobiome的“win”文件夹中以备将来使用。请注意,某些参考数据库可能最初以“.gz”格式压缩。如果这是您第一次下载它们,则需要使用“gunzip”命令对它们进行解压缩,然后再继续。一旦解压完成,以后就可以跳过此步骤。以下代码演示了如何解压代表性的16S / 18S / ITS RDP、SILVA和UNITES数据库(图 2C)。
gunzip -c ${db}/usearch/rdp_16s_v18.fa.gz > ${db}/usearch/rdp_16s_v18.fa
seqkit stat ${db}/usearch/rdp_16s_v18.fa
seqkit stat ${db}/usearch/unite.fa
Greengene数据库用于特征注释,默认解压会删除原始文件,“-c”指定输出, “>”写入一个新文件并重命名。
gunzip -c ${db}/gg/97_otus.fasta.gz > ${db}/gg/97_otus.fa
seqkit stat ${db}/gg/97_otus.fa
Reads合并与重命名
本节介绍如何使用VSEARCH软件合并和重命名双端测序数据文件。合并指将每个样本的正向和反向reads合并到一个文件中,而重命名可确保下游分析的命名一致(图 2D)。
合并和重命名过程
EasyAmplicon演示了合并和重命名的两种主要方法。
方法1(使用“for”循环):此方法适用于所有系统,遍历元数据文件中列出的样本对每对reads进行合并和重命名。
time for i in `tail -n+2 result/metadata.txt|cut -f1`;do
vsearch --fastq_mergepairs seq/${i}_1.fq.gz --reverse seq/${i}_2.fq.gz \
--fastqout temp/${i}.merged.fq --relabel ${i}.
done &
方法2(使用“rush”):此方法利用“rush”命令执行并行处理,潜在地提高了多核系统的速度。使用此方法之前,请确保您的系统支持“rush”,因为某些计算机并不支持,并且可能在执行过程中调度失败。如果是这种情况,请改用“for”循环)。
time tail -n+2 result/metadata.txt | cut -f 1 | \
rush -j 3 "vsearch --fastq_mergepairs seq/{}_1.fq.gz --reverse seq/{}_2.fq.gz \
--fastqout temp/{}.merged.fq --relabel {}."
运行此代码检查最后一个文件前10行中的样本名称:
head temp/`tail -n+2 result/metadata.txt | cut -f 1 | tail -n1`.merged.fq | grep ^@
注意:无论选择哪种方法,都请确保脚本或循环执行完成后再继续下一步。处理不完整可能会导致数据不一致。
重命名后整合序列
最后一步,通过运行代码将所有合并和重命名的文件合并到临时目录中名为“all.fq”的文件中cat temp/*.merged.fq > temp/all.fq。要检查合并文件的大小,请使用ls -lsh temp/all.fq。使用代码确保样本名称在点(.)之前head -n 6 temp/all.fq | cut -c1-60。这有助于避免后续分析中出现内存问题。
修剪引物并进行质量控制
本节介绍如何修剪引物序列并对合并和重命名的reads进行质量控制(图 2E)。此处假设引物长度左侧为29 bp,右侧为18 bp。请务必根据实际引物设计调整这些值。
time vsearch --fastx_filter temp/all.fq --fastq_stripleft 29 --fastq_stripright 18 --fastq_maxee_rate 0.01 --fastaout temp/filtered.fa
去冗余并选择OTU / ASV
本节介绍两个关键步骤:去冗余和OTU / ASV选择(图 2F)。去冗余过程会删除相同的序列,只保留单个副本及其丰度信息。这可以减小文件大小并加快进一步分析的速度。OTU / ASV选择步骤将相似序列分组为操作分类单元(OTU)或扩增子序列变体(ASV)。OTU代表具有高相似性的序列簇(通常为97%或更高),而ASV针对于单核苷酸准确性。
去冗余
运行命令vsearch --derep_fulllength temp/filtered.fa --minuniquesize 10 --sizeout --relabel Uni_ --output temp/uniques.fa。代码演示了如何使用VSEARCH去除重复序列。“--derep_fulllength”选项可识别并删除相同的序列。“--minuniquesize”设置最小丰度阈值以排除非常罕见的序列(视为噪音)。通常使用10这个值。“--sizeout”输出包含序列丰度信息的表。“--relabel Uni_”在序列名称中添加前缀以便更好地组织。该命令生成两个文件:(i)temp/uniques.fa(此文件包含去冗余序列),(ii)temp/uniques.fa.sizeout(此文件包含每个序列的丰度信息。)
笔记:“--minuniquesize”阈值可根据用户的数据和分析要求设定。较高的阈值会删除更多的低丰度序列。
聚类OTU或去噪ASV
本节讨论了对相似序列进行分组的两种主要方法,包括OTU聚类和ASV去噪。在VSEARCH中,两种方法都自带从头(De novo)去除嵌合体,从而消除PCR扩增过程中形成的人工序列。
方法1(OTU聚类):这种传统方法将相似度在97%以上的序列划分为OTU,适用于大型数据集或ASV规律不明显的情况:
usearch -cluster_otus temp/uniques.fa -minsize 10 \
-otus temp/otus.fa \
-relabel OTU_
方法2(ASV去噪):为了识别准确的生物序列并消除测序错误以获得精确的序列信息,请执行以下代码:
usearch -unoise3 temp/uniques.fa -minsize 10 -zotus temp/zotus.fa
修改序列名称:将“Zotu”更改为“ASV”,以便于识别:
sed 's/Zotu/ASV_/g' temp/zotus.fa > temp/otus.fa
head -n 2 temp/otus.fa
注意:ASV去噪的处理时间因数据集大小而异。对于大型数据集,或者不需要精确的序列信息时,请使用OTU聚类。ASV去噪更适合用于需要高精度序列数据的研究(例如微生物群落多样性分析)。
基于参考的嵌合体检测
通常不建议去除嵌合体,因为参考数据库中缺乏丰度信息,这可能导致假阴性。而与之相对的是,从头开始的嵌合体检测又要求亲本丰度至少是嵌合体的16倍以防止假阴性。由于已知的序列不会被去除,因此只有参考数据库越大才越能够将假阴性率降到最低。
mkdir -p result/raw
方法1:使用VSEARCH结合RDP去除嵌合体(速度快但容易出现假阴性)。下载并提取SILVA数据库(将rdp_16s_v18.fa替换为silva_16s_v123.fa)。这个过程非常慢,但理论上可以提供更好的结果。
vsearch --uchime_ref temp/otus.fa \
-db ${db}/usearch/rdp_16s_v18.fa \
--nonchimeras result/raw/otus.fa
如果您使用的是Windows,VSEARCH结果可能会添加行尾(^M),您需要将其删除。但是如果您使用的是Mac,则无需执行此命令:
sed -i 's/\r//g' result/raw/otus.fa
方法2:此方法提供了一种不进行嵌合体去除的替代方法。如果您已经处理了嵌合体,或者对于需要未过滤OTU的特定下游分析,这种方法会很有用。
cp -f temp/otus.fa result/raw/otus.fa
特征表构建与过滤
本节介绍如何生成特征表,该表总结了每个样本中发现的特征(OTU或ASV)的丰度,还包含基于分类学分类的物种注释和筛选的可选步骤(图 2G)。
根据样本大小生成特征表
生成特征表的方法的选择取决于数据集的大小,有两种方法可以生成特征表。
方法1:此方法使用USERACH生成特征表。对于小样本来说,它很快(< 30),但对于较大的数据集来说效率低下。
time usearch -otutab temp/filtered.fa
-otus result/raw/otus.fa
-threads 4
-otutabout result/raw/otutab.txt
方法2:此方法利用VSEARCH生成特征表,总结每个OTU的reads计数,特别适合处理大型数据集。
time vsearch --usearch_global temp/filtered.fa
--db result/raw/otus.fa
--id 0.97 --threads 4
--otutabout result/raw/otutab.txt
所选方法、数据集大小和可用的计算资源共同决定特征表的生成时间。VSEARCH提供的多线程功能可以提高处理速度。输出的特征表(result/raw/otutab.txt)将包含每个样本中每个特征(OTU / ASV)的丰度信息。
物种注释——去除质体和非细菌 / 古菌(可选)
本节介绍用物种信息注释特征和基于系统分类筛选数据(图 2H)。对于物种注释,使用RDP或SILVA等参考数据库为特征分配潜在分类。虽然RDP更快,但SILVA数据库更全面。
vsearch --sintax result/raw/otus.fa \
--db ${db}/usearch/rdp_16s_v18.fa \
--sintax_cutoff 0.1 \
--tabbedout result/raw/otus.sintax
head result/raw/otus.sintax | cat -A
sed -i 's/\r//' result/raw/otus.sintax
方法1:基于分类学筛选。基于注释,过滤特征表和序列,排除非细菌/古细菌序列(例如,来自质体或线粒体的序列)。代码演示了使用R脚本根据特征表(result/raw/otutab.txt)和物种注释(result/raw/otus.sintax)进行筛选。(要筛选真菌ITS数据,请改用otutab_filter_nonFungi.R)
wc -l result/raw/otutab.txt
Rscript ${db}/script/otutab_filter_nonBac.R \
--input result/raw/otutab.txt \
--taxonomy result/raw/otus.sintax \
--output result/otutab.txt\
--stat result/raw/otutab_nonBac.stat \
--discard result/raw/otus.sintax.discard
筛选后的特征表行数:
wc -l result/otutab.txt
过滤特征表对应序列:
cut -f 1 result/otutab.txt | tail -n+2 > result/otutab.id
usearch -fastx_getseqs result/raw/otus.fa \
-labels result/otutab.id -fastaout result/otus.fa
过滤特征表对应序列注释:
awk 'NR==FNR{a[$1]=$0}NR>FNR{print a[$1]}'\
result/raw/otus.sintax result/otutab.id \
> result/otus.sintax
方法2:如果您认为前面步骤中的初步筛选已经足够,则此方法提供了一个选项可以跳过对生成的特征表进行额外过滤。
cp result/raw/otu* result/
要对OTU表进行简单统计,可运行下列代码:
usearch -otutab_stats result/otutab.txt \
-output result/otutab.stat
cat result/otutab.stat
等量抽样标准化
这一步解决了样本间reads计数的规范化问题,以解释测序深度的潜在差异(图 2I)。提供的R脚本利用vegan包执行该步骤,输出文件“otutab_rare.txt”和“vegan.txt”,可用于下游Alpha多样性分析。
mkdir -p result/alpha
Rscript ${db}/script/otutab_rare.R --input result/otutab.txt \
--depth 10000 --seed 1 \
--normalize result/otutab_rare.txt \
--output result/alpha/vegan.txt
usearch -otutab_stats result/otutab_rare.txt \
-output result/otutab_rare.stat
cat result/otutab_rare.stat
注意:由于此流程是为端到端分析量身定制的,因此本节中生成的文件将用于在后续分析中生成Alpha多样性图。图2详述了所有上述步骤。
分析(从大表到小表)
视频链接教程:https://youtu.be/R7wj3D9B6xU
特征表是微生物组分析中数据处理步骤中获得的关键结果。它通过汇总每个样本映射到特定OTU的reads数(序列)来压缩测序数据。这些表对于计算样本内(Alpha多样性)和样本间(Beta多样性)的多样性至关重要。此外,分类信息可让研究人员探索特定的微生物群(例如属、科)并识别与某些条件相关的生物标志物(图 3)。特征表还可以进行进一步分析,例如差异丰度测试,它可以识别与特定条件或状态显着相关的微生物。
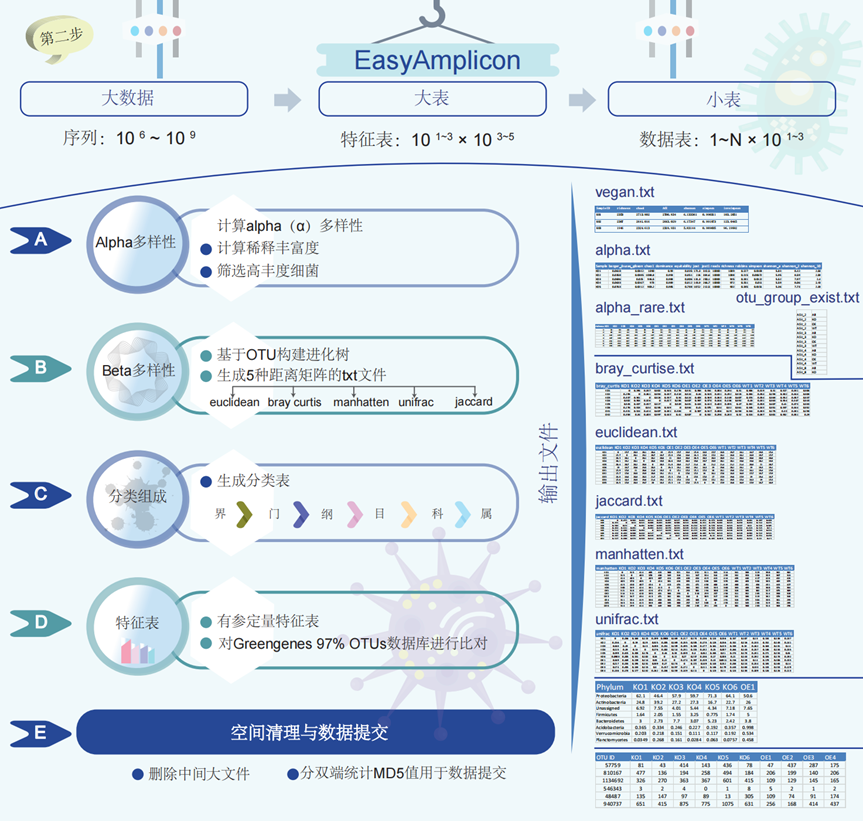
图3. EasyAmplicon分类多样性分析流程步骤
(A)Alpha多样性涉及分析样本中的物种多样性以总结微生物多样性。输出包括alpha.txt、alpha_rare.txt和OTU_group_exist文件。(B)Beta多样性涉及分析样本之间的物种多样性,以总结不同样本之间的微生物多样性。输出包括距离矩阵的文本文件:bray_curtis.txt、euclidean.txt、jaccard.txt、manhattan.txt和unifrac.txt。这些文件包含距离矩阵,用于测量样本之间在微生物群落方面的差异。(C)分类组成:此步骤将微生物分为分类群,包括门、纲、目、科和属。(D)特征表:此表包含丰度和分类信息,并与Greengenes数据库(97% OTU)进行了比对。(E)空间清理和数据提交是最后一步,删除临时文件并准备数据以提交到公共存储库,包括计算双端数据的MD5值。
Alpha(α)多样性分析
计算Alpha多样性指数
此步骤重点计算Alpha多样性,这是一种量化样本内物种丰富度和均匀度的指标(图 3A)。USEARCH是此流程中包含的一个多功能工具,可以计算14种不同的Alpha多样性指数。有关这些指数的详细描述,请参阅USEARCH手册(http://www.drive5.com/usearch/manual/alpha_metrics.html),并在R中运行给定的代码,该代码使用包含Alpha多样性计算数据的“otutab_rare.txt”,并生成输出文件“alpha.txt”。
usearch -alpha_div result/otutab_rare.txt -output result/alpha/alpha.txt
稀释丰富度分析
该流程采用稀释曲线法,该方法涉及以不同百分比(通常在1%到100%之间)对测序数据进行二次采样。为了评估微生物群落的多样性,同时补偿样本间测序深度的差异,请执行稀释分析。该分析估计了在特定测序深度处识别的OTU的数量。执行以下代码,该代码利用USEARCH生成一个名为“alpha_rare.txt”的文件,其中包含稀释丰富度数据:
usearch -alpha_div_rare result/otutab_rare.txt -output result/alpha/alpha_rare.txt \
-method without_replacement
head -n2 result/alpha/alpha_rare.txt
sed -i "s/-/\t0.0/g" result/alpha/alpha_rare.txt
“without_replacement”选项确保二次采样时每个OTU都有相同的被抽中的概率,从而减轻因测序深度不均匀而造成的偏差。
高丰度细菌过滤(可选)
此步骤允许用户从样本数据中过滤出高丰度的细菌,这有助于将下游分析的重点放在可能被优势物种掩盖的数量较少的分类群上。根据metadata.txt文件中的实验设计调整组列名,使用特征表(result/otutab.txt)和元数据文件(metadata.txt)作为输入文件,输出一个修改后的特征表,其中包含分组方法。使用代码Rscript ${db}/script/otu_mean.R -h,其中“-h”选项显示脚本的帮助,提供参数说明。参数“scale”决定是否进行缩放,“zoom”将总和标准化,“all”输出所有样本的平均值,“type”指定计算类型为“mean”或“sum”。
Rscript ${db}/script/otu_mean.R --input result/otutab.txt \
--metadata result/metadata.txt \
--group Group --thre 0 \
--scale TRUE --zoom 100 --all TRUE --type mean \
--output result/otutab_mean.txt
输出包括总体平均值和每个组的平均值。
head -n3 result/otutab_mean.txt
通过平均丰度阈值 > 0.1%(也可选择0.5%或0.05%)筛选得到的各组OTU组合。
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {for(i=3;i<=NF;i++) a[i]=$i; print "OTU","Group";} \
else {for(i=3;i<=NF;i++) if($i>0.1) print $1, a[i];}}' \
result/otutab_mean.txt > result/alpha/otu_group_exist.txt
head result/alpha/otu_group_exist.txt
cut -f 2 result/alpha/otu_group_exist.txt | sort | uniq -c
注意:或者,浏览此链接http://ehbio.com/test/venn/,在线分析不同丰度下,各组间的OTU/ASV计数。该工具还可以生成维恩图或网络图,可视化各组共有和特有的图像。
Beta(β)多样性
Beta多样性是一个生态学概念,特指不同类群或生态位之间的物种组成差异。在宏基因组学领域,通常用散点图显示样本组之间的Beta多样性,常用的分析方法包括主成分分析(PCA)、主坐标分析(PCoA/MDS)和限制性主坐标分析 (CPCoA/CCA/RDA)。PCoA的计算基于距离矩阵,其中unifrac距离矩阵依赖于系统发育树(图 3B)。
执行mkdir -p result/beta/命令,在result文件夹下创建一个名为“beta”的目录,用于存放Beta多样性输出文件。运行usearch -cluster_agg result/otus.fa -treeout result/otus.tree,该命令利用USEARCH软件根据存储在“result/otus.fa”中的OTU序列构建系统发育树。生成的树将被保存为“result/otus.tree”。下一个命令使用USEARCH根据OTU表(“result/otutab_rare.txt”)和构建的系统发育树(“result/otus.tree”)生成五个不同的距离矩阵,并以前缀“result/beta/”保存,后面跟着特定的距离度量名称(例如,“bray_curtis”,“euclidean”等)。
usearch -beta_div result/otutab_rare.txt -tree result/otus.tree \
-filename_prefix result/beta/
物种注释汇编
为了汇总物种注释分类,将OTU对应的物种注释重新格式化为两列结构,需要去除SINTAX中的置信值,只保留物种注释,“:”替换为“_”,并删除所有引号:cut -f 1,4 result/otus.sintax \
|sed 's/\td/\tk/;s/:/__/g;s/,/;/g;s/"//g' \
> result/taxonomy2.txt
head -n3 result/taxonomy2.txt
运行以下代码生成一个物种表,其中OTU/ASV中的空白用“未分配”填充。
awk 'BEGIN{OFS=FS="\t"}{delete a; a["k"]="Unassigned";a["p"]="Unassigned";a["c"]="Unassigned";a["o"]="Unassigned";a["f"]="Unassigned";a["g"]="Unassigned";a["s"]="Unassigned";\
split($2,x,";");for(i in x){split(x[i],b,"__");a[b[1]]=b[2];} \
print $1,a["k"],a["p"],a["c"],a["o"],a["f"],a["g"],a["s"];}' \
result/taxonomy2.txt > temp/otus.tax
sed 's/;/\t/g;s/.__//g;' temp/otus.tax|cut -f 1-8 | \
sed '1 s/^/OTUID\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' \
> result/taxonomy.txt
head -n3 result/taxonomy.txt
接下来,执行以下命令来计算不同层级的分类单元数量:门、纲、目、科和属。分别使用参数p、c、o、f 和g(图 3C)。这将有助于对不同分类级别的微生物群落进行分类和量化,从而获取有关其组成的详细视图。
mkdir -p result/tax
for i in p c o f g;do
usearch -sintax_summary result/otus.sintax \
-otutabin result/otutab_rare.txt -rank ${i} \
-output result/tax/sum_${i}.txt
done
sed -i 's/(//g;s/)//g;s/\"//g;s/\#//g;s/\/Chloroplast//g' result/tax/sum_*.txt
要列出所有文件,请运行此代码:
wc -l result/tax/sum_*.txt
head -n3 result/tax/sum_g.txt
注意:这里生成的输出文件sum_p.txt将在接下来的步骤中用于构建堆叠直方图,c sum_c.txt用于和弦图。
有参定量特征表
为了生成有参定量特征表,将序列与Greengenes 97% OTU数据库比对,用于PICRUSt/Bugbase功能预测(图3D)。首先,创建一个目录来存储比对结果:mkdir -p result/gg/。根据大小,可以使用多种方法来执行此步骤。
方法1:使用USEARCH比对,此方法提供更快的处理速度。但是请考虑文件大小限制,尤其是大型数据集。示例演示了使用4个线程进行比对,比对率为80.0%。处理时间和内存使用量因硬件规格而异。
usearch -otutab temp/filtered.fa -otus ${db}/gg/97_otus.fa -otutabout result/gg/otutab.txt -threads 4
方法2 :VSEARCH比对精度更高但处理时间更长。它利用并行化来提高性能,尤其是在使用24-96个线程的情况下。注释的示例演示使用12个线程,实现了81.04%的对齐率。
vsearch --usearch_global temp/filtered.fa --db ${db}/gg/97_otus.fa --otutabout result/gg/otutab.txt --id 0.97 --threads 12
要查找统计数据,请运行以下代码:
usearch -otutab_stats result/gg/otutab.txt -output result/gg/otutab.stat
cat result/gg/otutab.stat
空间清理和数据提交
此步骤包括分析后的空间清理和准备数据的提交(图 3E)。执行rm -rf temp/*.fq删除临时文件以释放存储空间。
通过分双端统计MD5值用于数据提交:
cd seq
md5sum *_1.fq.gz > md5sum1.txt
md5sum *_2.fq.gz > md5sum2.txt
paste md5sum1.txt md5sum2.txt | awk '{print $2"\t"$1"\t"$4"\t"$3}' \
| sed 's/*//g' > ../result/md5sum.txt
rm md5sum*
cd ..
cat result/md5sum.txt
图表统计与可视化
视频教程链接:https://youtu.be/n13M8p_IrXk
继续进行R下的多样性(Alpha和Beta)和物种组成分析,以生成Alpha多样性、Beta多样性和物种组成分类的箱线图。可使用位于EasyMicrobiome(db)文件夹中的R脚本执行各种微生物组分析任务。
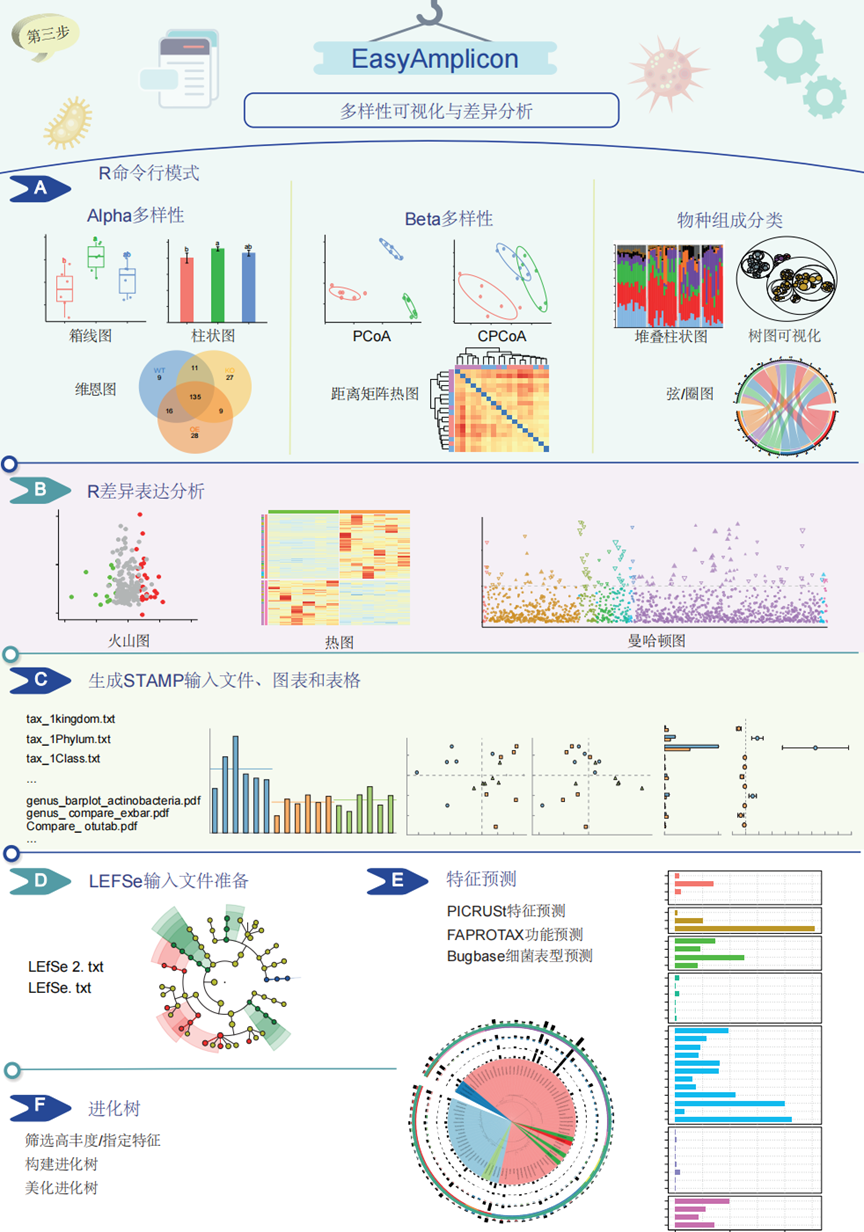
图4. EasyAmplicon流程分析提供了多种功能来分析扩增子测序数据
(A)该流程通过生成箱线图、直方图和维恩图等Alpha多样性可视化图来深入研究样本中的物种多样性。Beta多样性分析探索不同样本中微生物群落之间的差异;该流程通过距离矩阵热图、主坐标分析(PCoA)和限制性主坐标分析 (CPCoA)图分析Beta多样性。此外,该流程通过弦图、堆叠柱状图以及物种组成和分类的树状图来研究样本微生物群落的分类组成。(B)差异表达分析可视化包括热图、火山图和曼哈顿图。(C-D)除了这些核心分析之外,该流程还可以通过为STAMP和LEfSe生成兼容的输入文件进行深入分析,从而与第三方软件无缝集成。(E)EasyAmplicon还具备深入分析特定特征的功能,包括使用PICRUSt、FAPROTAX和BugBase等工具预测细菌表型,(F)以及使用iQTree软件构建进化树。
Alpha多样性绘图
EasyAmplicon 流程提供生成各种Alpha多样性图的功能,可深入了解每个样本中微生物群落的丰富度和均匀度。这些图包括Alpha柱状图(通常显示观察到的OTU数量)、Alpha箱线图(可视化OTU计数分布)和Alpha稀释曲线(估计同一测序深度的多样性)。都是通过数据文件(例如alpha.txt、vegan.txt、alpha_rare.txt、otu_group_exist.txt)与ggplot2和vegan等R包来生成的(图 4A)。
箱线图
运行以下命令绘制Alpha箱线图:
Rscript ${db}/script/alpha_boxplot.R -h
Rscript ${db}/script/alpha_boxplot.R --alpha_index richness \
--input result/alpha/vegan.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 89 --height 59
使用循环绘制6种常用指数:
for i in `head -n1 result/alpha/vegan.txt|cut -f 2-`;do
Rscript ${db}/script/alpha_boxplot.R --alpha_index ${i} \
--input result/alpha/vegan.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 89 --height 59
done
mv alpha_boxplot_TukeyHSD.txt result/alpha/
此外,要绘制Alpha多样性柱状图并计算标准差,请运行以下命令:
Rscript ${db}/script/alpha_barplot.R --alpha_index richness \
--input result/alpha/vegan.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 89 --height 59
稀释曲线
稀释曲线说明了组测序深度平均值与特征标准误差(OTU或ASV)计数。输入文件alpha_rare.txt,指定实验设计和分组,设置输出目录,并定义图像尺寸(宽度和高度)。为了绘制曲线,请运行以下代码:
Rscript ${db}/script/alpha_rare_curve.R \
--input result/alpha/alpha_rare.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 120 --height 59
维恩图
方法1:要绘制Alpha多样性维恩图,需输入文件otu_group_exist.txt,该文件包含两列:ID和group。命令行如下:其中“-f”用于输入文件,“-a/b/c/d/g”用于组名,“-w/u”用于宽度和高度(单位为英寸),“-p”用于输出文件名(默认值与输入相同)(图 4A)。运行以下代码比较三个组:
bash ${db}/script/sp_vennDiagram.sh \
-f result/alpha/otu_group_exist.txt \
-a WT -b KO -c OE \
-w 3 -u 3 \
-p WT_KO_OE
运行以下代码比较四个组:
bash ${db}/script/sp_vennDiagram.sh \
-f result/alpha/otu_group_exist.txt \
-a WT -b KO -c OE -d All \
-w 3 -u 3 \
-p WT_KO_OE_All
方法2(使用R Markdown(Rmd)绘制Alpha箱线图):本节将引导您使用位于EasyAmplicon主文件夹的“result”目录或EasyMicrobiome的脚本目录。在RStudio中打开Diversity.Rmd文件以运行代码并生成可视化。在Rmd文件的实验设计部分,仔细检查与您的实验相关的元数据。这涉及验证分组变量(group)、图像尺寸(width和height),以及确认用于Alpha多样性计算的文件和索引类型,默认为“Richness”。有两种方法可以执行代码:首先,您可以逐行运行代码,这样您就可以在代码执行过程中监控每个变量的创建。或者,您可以使用RStudio中的右箭头按钮按顺序执行所有代码块,或者“Knit”整个文档,这将生成包含所有结果和计算的综合网页报告。生成的输出可以在“alpha”目录中找到。Alpha多样性(丰富度)的箱线图位于文件“alpha/alpha_boxplot_Richness.pdf”中。用户还可以在“alpha”目录中找到稀释曲线(如果包含),具体在“alpha/alpha_rarefaction_curve.pdf ”中。“#稀释曲线”部分中的说明将提供特定可视化图输入和输出文件位置的详细信息。
方法3:此外,用户可以访问ImageGP(https://www.bic.ac.cn/BIC/#/),在线绘制箱线图(图 S1-A)。首先在Excel中打开数据文件(metadata.txt和vegan.txt),然后将“group”和“richness”列复制到新工作表中。将这些组合数据粘贴到网站文本框中,并选择分组(group)和丰富度(Y 轴)。单击“PLOT ”以生成箱线图。同样,用户可以使用此链接绘制维恩图和稀释曲线(图 S1-A)。
Beta多样性
距离矩阵热图
距离矩阵样本的微生物组成来表示样本之间的差异。它在微生物组分析中起着至关重要的作用,例如对相似样本进行聚类。要使用Bray-Curtis作为示例绘制距离矩阵热图,您可以运行以下代码:
bash ${db}/script/sp_pheatmap.sh \
-f result/beta/bray_curtis.txt \
-H 'TRUE' -u 6 -v 5
要在第2列和第4列添加分组注释(例如基因型和位置),请运行以下代码:
cut -f 1-2 result/metadata.txt > temp/group.txt
接下来绘制热图:
bash ${db}/script/sp_pheatmap.sh \
-f result/beta/bray_curtis.txt \
-H 'TRUE' -u 6.9 -v 5.6 \
-P temp/group.txt -Q temp/group.txt
其中“-f”是输入文件,“-H”是聚类(TRUE / FALSE),“-u / v”是宽度和高度(英寸),“-P”为文件添加行注释,“-Q”添加列注释(图 4A)。
主坐标分析PCoA
本节介绍如何执行主坐标分析(PCoA)来可视化数据中的Beta多样性。主要有两种方法:
方法1:此方法使用R脚本”beta_pcoa.R”,利用bray_curtis.txt和metadata.txt作为输入文件并输出bray_curtis.cpcoa.pdf。
Rscript ${db}/script/beta_pcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --label FALSE --width 89 --height 59 \
--output result/beta/bray_curtis.pcoa.pdf
要使用参数“--label”添加示例标签,请运行以下脚本:
Rscript ${db}/script/beta_pcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --label TRUE --width 89 --height 59 \
--output result/beta/bray_curtis.pcoa.label.pdf
mv beta_pcoa_stat.txt result/beta/
若要查看组间两两比较P值,请运行以下代码(概率事件每次略有不同):
cat result/beta/beta_pcoa_stat.txt
方法2:使用“Diversity.Rmd”绘制PCoA:使用R studio打开“Diversity.Rmd”文件,检查并运行“#β diversity多样性——##PCoA主坐标轴分析”。Knit输出文档生成结果和计算过程,包含beta/pcoa_bray_curtis.pdf和beta_pcoa_stat.txt(包含组间两两比较P值)两个文件。
方法3:同样,用户也可以使用在线工具http://www.bic.ac.cn/ImageGP/或https://www.bic.ac.cn/BIC/#/在线绘制PCoA图和热图。(图 S1-A)。
限制性主坐标分析CPCoA
本节介绍限制性主坐标分析(CPCoA),可探究环境因素或其他变量如何影响Beta多样性。执行CPCoA有两种主要方法:
方法1:此方法利用提供的R脚本“beta_cpcoa.R”。脚本参数与“beta_pcoa.R”脚本类似:
Rscript ${db}/script/beta_cpcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --output result/beta/bray_curtis.cpcoa.pdf \
--width 89 --height 59
使用--label TRUE参数添加样本标签:
Rscript ${db}/script/beta_cpcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --label TRUE --width 89 --height 59 \
--output result/beta/bray_curtis.cpcoa.label.pdf
方法2:使用“Diversity.Rmd”脚本可视化CPCoA图。打开“Diversity.Rmd”文件,找到“#β diversity多样性——## Constrained有监督PCoA/CCA”部分。修改脚本中的文件路径为实际Beta多样性矩阵位置(可能位于“result/beta”目录中)。文件路径调整后,“knit”R Markdown文档生成一份全面的网页报告,包含PCoA图和其他相关结果,以及一个名为“beta/cpcoa_bray_curtis.pdf”的附加文件,展示含有Bray-Curtis距离的CPCoA图。
方法3:虽然提供的脚本支持Bray-Curtis距离,但如果用户想要使用其他距离算法,请考虑使用在线工具http://www.bic.ac.cn/ImageGP/(图 S1-A)。此工具提供了十多个非进化距离选项,但无法计算与Unifrac相关的距离。
物种组成分类
用户可以通过多种可视化方式(如堆叠条形图stackplots、圈图chord diagrams和树状图)(图 4A)探索不同层级微生物群落的分类组成。每种图都有两种绘制方法。
堆叠柱状图
方法1:以门(p)水平为例,结果包括.sample.pdf和.group.pdf两种文件。输入文件是先前生成的sum_p.txt。
Rscript ${db}/script/tax_stackplot.R \
--input result/tax/sum_p.txt --design result/metadata.txt \
--group Group --color ggplot --legend 7 --width 89 --height 59 \
--output result/tax/sum_p.stackplot
可以使用ggplot manual(22)、Paired(12)或Set3(12)修改颜色。
Rscript ${db}/script/tax_stackplot.R \
--input result/tax/sum_p.txt --design result/metadata.txt \
--group Group --color Paired --legend 12 --width 181 --height 119 \
--output result/tax/sum_p.stackplotPaired
批量绘制包含门(p),纲(c),目(o),科(f),属(g)五个分类水平的输入数据:
for i in p c o f g; do
Rscript ${db}/script/tax_stackplot.R \
--input result/tax/sum_${i}.txt --design result/metadata.txt \
--group Group --output result/tax/sum_${i}.stackplot \
--legend 8 --width 89 --height 59; done
方法2(可选):通过.Rmd文件绘制堆叠直方图:用R studio打开“Diversity.Rmd”,检查“#物种组成——堆叠柱状图”段落参数,调整分类水平、输入输出文件位置和图片大小,点击命令即可运行。
弦/圈图
以纲(c)为例,运行以下代码绘制出前5组:
i=c
Rscript ${db}/script/tax_circlize.R \
--input result/tax/sum_${i}.txt --design result/metadata.txt \
--group Group --legend 5
结果位于当前目录“circlize_pdf”(随机颜色)和“circlize_legend.pdf”(指定颜色和图例)。要移动文件并改名使之与分类级一致:
mv circlize.pdf result/tax/sum_${i}.circlize.pdf
mv circlize_legend.pdf result/tax/sum_${i}.circlize_legend.pdf
方法2:运行.Rmd文件绘制弦/圈图。打开R studio的“Diversity.Rmd”并检查“## Circlize plot弦图”段落参数,调整分类水平、图例数量和分组列名。运行段落并在文件“circlize.pdf”和“circlize_legend.pdf”中查看结果。
树形图可视化
要可视化描述物种关系的分层树状图,请运行以下代码。通过输入特征表和物种注释来生成树状图,一定要指定特征的数量和图像的尺寸。
Rscript ${db}/script/tax_maptree.R \
--input result/otutab.txt --taxonomy result/taxonomy.txt \
--output result/tax/tax_maptree.pdf \
--topN 100 --width 183 --height 118
R差异分析
视频教程链接:https://youtu.be/FsdYNQ5DtHg
差异比较
这一步的重点是识别组间的显著不同的特征。为了分析差异,用户必须指定输入数据,定义比较组,设置参数,进行统计检验并生成火山图、热图、曼哈顿图、特征图和三元图(图 4B)。为此,需要在result文件夹中创建一个名为“compare”的目录,运行以下代码:mkdir -p result/compare/。使用输入特征表(otutab.txt)和元数据(metadata.txt)指定分组列名、比较组和丰度。选择edgeR方法,p值阈值为0.05,FDR阈值为0.2并制定输出目录,运行以下代码:
compare="KO-WT"
Rscript ${db}/script/compare.R \
--input result/otutab.txt --design result/metadata.txt \
--group Group --compare ${compare} --threshold 0.1 \
--method edgeR --pvalue 0.05 --fdr 0.2 \
--output result/compare/
故障提示:Error in file(file, ifelse(append, "a", "w"))。解决方法:提示输出目录不存在,新建目录即可。
差异丰度结果的可视化与分析
EasyAmplicon可通过绘制各种可视化图像来展示不同的丰度结果。
火山图
为了绘制火山图(图 4B)并可视化差异倍数和统计显著性之间的关系,运行以下代码:
Rscript ${db}/script/compare_volcano.R \
--input result/compare/${compare}.txt \
--output result/compare/${compare}.volcano.pdf \
--width 89 --height 59
热图
要绘制显示样本和组间特征丰度模式的热图,请运行以下代码,该代码筛选列数、指定元数据和分组、物种注释、图形大小和字体大小(图 4B)。
bash ${db}/script/compare_heatmap.sh -i result/compare/${compare}.txt -l 7 \
-d result/metadata.txt -A Group \
-t result/taxonomy.txt \
-w 8 -h 5 -s 7 \
-o result/compare/${compare}
曼哈顿图
本节介绍如何生成曼哈顿图,以可视化不同组间物种丰度的显著差异(图 4B)。这些图有助于识别区分样本的关键特征:
bash ${db}/script/compare_manhattan.sh -i result/compare/${compare}.txt \
-t result/taxonomy.txt \
-p result/tax/sum_p.txt \
-w 183 -v 59 -s 7 -l 10 \
-o result/compare/${compare}.manhattan.p.pdf
其中,“-i”表示差异比较结果,“-t”表示物种注释,“-p”表示图例,“-w”表示宽度,“-v”表示高度,“-s”表示大小,“-l”表示图例最大值。若要显示详细信息,请使用以下代码切换到c类和L类:
bash ${db}/script/compare_manhattan.sh -i result/compare/${compare}.txt \
-t result/taxonomy.txt \
-p result/tax/sum_c.txt \
-w 183 -v 59 -s 7 -l 10 -L Class \
-o result/compare/${compare}.manhattan.c.pdf
要显示完整的图例,请运行以下代码:
bash ${db}/script/compare_manhattan.sh -i result/compare/${compare}.txt \
-t result/taxonomy.txt \
-p result/tax/sum_c.txt \
-w 183 -v 149 -s 7 -l 10 -L Class \
-o result/compare/${compare}.manhattan.c.legend.pdf
注意:或者,用户可以使用ImageGP(http://www.bic.ac.cn/ImageGP/)绘制火山图、热图和曼哈顿图(图 S1-A)。
单个特征的绘制
这一步的重点是可视化和分析在前面步骤中识别的差异丰富序列变体(ASV)和操作分类单元(OTU)。运行下列代码,筛选差异ASV,按KO组丰度降序排列,并展示前10名ID:
awk '$4<0.05' result/compare/KO-WT.txt | sort -k7,7nr | cut -f1 | head
故障提示:如果使用awk命令后没有返回任何结果(空输出),则可能表明数据中缺乏差异丰富的ASV(p值> 0.05)。您可以通过调整显著性阈值(0.05)或比较组的潜在问题以调查数据。
差异OTU分析:为了显示差异OTU详细信息,运行以下命令:
Rscript ${db}/script/alpha_boxplot.R --alpha_index ASV_2 --input result/otutab.txt --design result/metadata.txt --transpose TRUE --scale TRUE --width 89 --height 59 --group Group --output result/compare/feature_
代码Rscript ${db}/script/alpha_boxplot.R将启动${db}/script目录下一个名为alpha_boxplot.R的脚本。该脚本将生成一个箱线图,比较“metadata.txt”文件中定义的不同组之间特定Alpha多样性指数(ASV_2)的丰度,脚本中的其他参数设置输出格式和尺寸。
故障提示:Error in data.frame(..., check.names = FALSE) :此错误表明箱线图数据中的行数有问题。如果“otutab.txt”或“metadata.txt”文件针对每个样品的行数不同,则可能出现这种情况。检查数据处理或过滤步骤中的不一致处,确保两个文件的样本信息互相匹配。
属级分析:要按平均属丰度降序对列进行排序,请运行以下命令:
csvtk -t sort -k All:nr result/tax/sum_g.txt | head
此代码利用“csvtk”工具对位于“result/tax”目录中的“sum_g.txt”文件进行排序。“-t”标志指定制表符分隔的文件,“-k All:nr”按降序对所有列进行数字排序,“head”显示最上面的行(显示最丰富的属)。要进一步探索属性级别差异的细节,请运行以下代码:
Rscript ${db}/script/alpha_boxplot.R --alpha_index Lysobacter \
--input result/tax/sum_g.txt --design result/metadata.txt \
--transpose TRUE \
--width 89 --height 59 \
--group Group --output result/compare/feature_
故障提示:如果根据属丰度数据生成的箱线图不能提供有效信息(细节不足),可能是由于属的数量太多。可以考虑过滤数据,将重点放在前几个最丰富的属上,或者使用柱状图等其他可视化方法。
使用STAMP进行差异丰度分析
视频教程链接:https://youtu.be/PptGLAI93eE
STAMP是一个用于分析微生物组数据的独立软件,本节概述了如何使用STAMP进行差异丰度分析(图4C)。
生成输入文件
该步骤涉及STAMP数据的格式化,以及生成差异丰度结果的可视化。
Rscript ${db}/script/format2stamp.R -h
mkdir -p result/stamp
Rscript ${db}/script/format2stamp.R --input result/otutab.txt --taxonomy result/taxonomy.txt --threshold 0.01 --output result/stamp/tax
输出结果位于results/stamp目录中。用户可以在STAMP软件中打开这些文件。结果包括名为tax_1至tax_8的文件。tax_1至tax_7是界、门、纲、目、科、属和种的分类摘要,tax_8是过滤后的OTU表。其中,例如0.01代表平均过滤丰度。此流程还提供了通过format2stamp.Rmd生成输入文件的替代方法,位于EasyAmplicon主文件夹的结果目录中或EasyMicrobiome数据库文件夹的脚本目录中。要使用format2stamp.Rmd,用户必须在结果目录中拥有“otutab.txt”和“taxonomy.txt”文件。接下来,使用R studio打开“format2stamp.Rmd”。在运行代码之前,请检查并调整代码参数。单击RStudio中的“Knit”按钮执行代码。默认情况下,结果将保存在result/stamp目录中。
绘制扩展柱状图和表格
要进行两组比较(例如KO-WT),建议使用Welch 's t检验+扩展误差条/热图。将ASV (result/otutab.txt)替换为属(result/tax/sum_g.txt),并运行如下命令:compare="KO-WT"
Rscript ${db}/script/compare_stamp.R \
--input result/stamp/tax_5Family.txt --metadata result/metadata.txt \
--group Group --compare ${compare} --threshold 0.1 \
--method "t.test" --pvalue 0.05 --fdr "none" \
--width 189 --height 159 \
--output result/stamp/${compare}
还可以通过“compareStamp.Rmd”作为可选方法,该方法可以在EasyAmplicon主文件夹的结果目录中或EasyMicrobiome文件夹的脚本目录中找到。此文件包含用户可以根据需要查看和调整的代码,包括输入和输出文件的名称和位置或特定阈值。对设置满意后,单击RStudio中的“Knit”按钮执行代码,生成所需的输入文件。默认情况下,结果将放置在“result/compare”目录中。
注意:多组比较建议采用PCA、ANOVA + FDR +单特征箱线图。
生成LEfSe输入文件
视频教程链接:https://youtu.be/VXJvdidyFlU
LEfSe分析用于识别组间存在显著差异的微生物特征。本节概述了用于生成LEfSe分析输入文件的两种方法(图 4D)。
方法1:在此方法中,运行以下命令生成输入文件。其中,threshold参数调节筛选丰度,控制图中分支数量。
mkdir -p result/lefse
Rscript ${db}/script/format2lefse.R --input result/otutab.txt --taxonomy result/taxonomy.txt --design result/metadata.txt --group Group --threshold 0.4 --output result/lefse/LEfSe
方法2(可选):或者, 用户可以通过使用format2lefse.Rmd文件生成输入文件。结果包含三个文件:otutab.txt、metadata.txt、taxonomy.txt。要继续分析的话请到db/script目录,找到format2lefse.Rmd文件,然后使用RStudio打开它。运行代码将生成两个输入文件:LEfSe.txt和LEfSe2.txt。为了进行全面的LEfSe分析和可视化,我们推荐两个在线工具。首先,使用ImageGP在线服务器(https://www.bic.ac.cn/BIC/#/analysis?page=b%27MzY%3D%27)。只需提交您的LEfSe输入文件(LEfSe.txt),大约3分钟内,您就会收到高质量的可视化图(图 S1-B)。或者,LEfSe官网(https://huttenhower.sph.harvard.edu/lefse/)也提供在线LEfSe分析平台。
方法3:Linux下的LEfSe本地分析(可选),该部分将在下文“Linux下的分析”中进行说明。
特征预测分析
本节介绍如何使用PICRUSt和Bugbase分别预测微生物群落的功能和形态特征。此处的特征预测是指从扩增子测序数据生成的序列中预测或识别功能的过程。
PICRUSt特征预测
视频教程链接:https://youtu.be/bq9IkEJVAoQ
PICRUSt是一种专门设计用于根据标记基因数据(通常是16S rDNA基因序列)预测微生物群落功能能力(例如代谢途径)的分析工具(图 4E)。
PICRUSt 1.0
方法1(Windows本地运行):第一步是文件准备,即通过比对Greengenes数据库生成OTU表。这已经在上面完成(请参阅pipeline.sh/pipeline_en.sh,步骤9以供参考),生成“otutab.txt”文件。第二步是使用在线网络服务器ImageGP(https://www.bic.ac.cn/BIC/#/)上传gg/otutab.txt文件。上传前请确保将OUT ID 修改为“OTUID”。生成的结果有四个级别(图 S1-C),下载所有输出文件并保存在工作目录路径EasyAmplicon/result/picrust中(提前在结果目录中生成picrust文件夹)。获得的文件可以直接在STAMP中进行可视化,因为STAMP是整合和显示各级分析结果的强大工具。
现在,运行以下代码以从ImageGP下载的文件中删除多余的细节,以便使文件格式与后续分析保持一致。
cd ${wd}/result/picrust/
find . -type f -name "*all_level*" -exec bash -c 'mv "$0" "${0/*all_level/all_level}"' {} \;
cd ${wd}
接下来运行下面给出的代码:
l=L2
sed '/# Const/d;s/OTU //' result/picrust/all_level.ko.${l}.txt > result/picrust/${l}.txt
num=`head -n1 result/picrust/${l}.txt|wc -w`
paste <(cut -f $num result/picrust/${l}.txt) <(cut -f 1-$[num-1] result/picrust/${l}.txt) \
> result/picrust/${l}.spf
cut -f 2- result/picrust/${l}.spf > result/picrust/${l}.mat.txt
awk 'BEGIN{FS=OFS="\t"} {print $2,$1}' result/picrust/${l}.spf | sed 's/;/\t/' | sed '1 s/ID/Pathway\tCategory/' \
> result/picrust/${l}.anno.txt
为了可视化KEGG各层级柱状图,执行以下代码生成输入文件。compare="KO-WT"
Rscript ${db}/script/compare.R --input result/picrust/${l}.mat.txt --design result/metadata.txt --group Group --compare ${compare} --threshold 0 --method wilcox --pvalue 0.05 --fdr 0.2 --output result/picrust/
然后,基于KEGG预测函数绘制微生物群落丰度分布图,将结果${compare}过滤成“.txt 文件”。执行以下代码生成柱状图:
Rscript ${db}/script/compare_hierarchy_facet.R --input result/picrust/${compare}.txt --data MeanA --annotation result/picrust/${l}.anno.txt --output result/picrust/${compare}.MeanA.bar.pdf
接下来,运行以下代码来生成描绘两组之间显著差异的柱状图,并根据更高的分类级别进行面划分:
Rscript ${db}/script/compare_hierarchy_facet2.R \
--input result/picrust/${compare}.txt \
--pvalue 0.05 --fdr 0.1 \
--annotation result/picrust/${l}.anno.txt \
--output result/picrust/${compare}.bar.pdf
方法2(本地操作-仅限于Linux系统,需安装picrust环境):首先启动Linux子系统并安装Conda。使用conda install picrust安装特征预测需要的软件环境。为了与下游分析兼容,请将您的OTU表转换为通用格式。此方法将在“Linux系统下的分析”部分详细说明。
FAPROTAX
视频教程链接:https://youtu.be/kAiMq08mLNs
FAPROTAX数据库(http://www.loucalab.com/archive/FAPROTAX)可基于16S rDNA基因扩增子数据预测潜在功能。它提供了一个免费的Python脚本(collapse_table.py),可将分类学概况(如OTU表)转换为潜在的功能概况。FAPROTAX十分全面,包含4,600多个分类群的7,600多个功能注释,涵盖80多种功能,以及代谢途径和生态相关过程,如硝化、反硝化和发酵。该资源将特定的原核生物分类群(例如属或种)映射到功能上,帮助研究人员揭示微生物群落的代谢潜力。
方法1:ImageGP作为一键式的在线网络平台,可使用FAPROTAX对微生物群落进行功能注释。将OTU表上传到ImageGP在线网络服务器(https://www.bic.ac.cn/BIC/#/),它将可以进行FAPROTAX分析(图 S1-D),提供全面的结果和可视化。
方法2(Linux系统下分析,可选):详细信息可在“Linux系统下的分析”部分中找到。这种方法为Linux经验丰富的用户提供了灵活选择。
Bugbase细菌表型预测
视频教程链接:https://youtu.be/OvkZxjOjwmE
BugBase是一个分析微生物组样本表型的工具。该网站可以根据OTU表和映射文件(Mapping files)预测和比较大量的信息,包括以下七个方面:革兰氏阳性、革兰氏阴性、生物膜形成、致病潜力、移动元件含量、氧利用和氧化胁迫耐受。研究中通常使用BugBase基于16S预测样品中革兰氏阳性/阴性、胁迫和致病菌的相对丰度。
方法1(Window本地运行):基于R 4.x更新代码和包。在pipeline.sh中指定软件目录作为Bugbase变量,输入gg OTU表、元数据,指定分组列名和输出目录。
cd ${wd}/result
bugbase=${db}/script/BugBase
rm -rf bugbase/
Rscript ${bugbase}/bin/run.bugbase.r -L ${bugbase} -i gg/otutab.txt -m metadata.txt -c Group -o bugbase/
方法2:或者,用户可以使用在线网络服务器通过ImageGP进行Bugbase分析(http://www.bic.ac.cn/ImageGP/index.php/Home/Index/BugBase.html) (图 S1-E)。此外,用户还可以访问Bugbase网站https://bugbase.cs.umn.edu/进行分析。但是,由于其容易出错,我们并不建议这样做。
方法3(Linux系统下分析,可选):Linux系统下Bugbase的安装和使用详细说明请参见“Linux系统下分析”部分。
高级系统发育树的构建
视频教程链接:https://youtu.be/c3cM-zwbChs
本节介绍如何使用各种方案构建和可视化系统发育树。通过执行以下代码,在“result”文件夹中创建一个名为“tree”的新文件夹来存储生成的文件(图 4F):
cd ${wd}
mkdir -p result/tree
cd ${wd}/result/tree
筛选高丰度/特定特征
方法1:基于丰度筛选特征。通常选择0.001或0.005,且OTU计数在30-150的范围内。使用代码tail -n+2 ../otutab_rare.txt | wc -l计算特征表中ASV的个数。然后运行usearch -otutab_trim ../otutab_rare.txt -min_otu_freq 0.002 -output otutab.txt命令,根据相对丰度阈值0.2%筛选高丰度的OTU,最后使用 tail -n+2 otutab.txt | wc -l计算筛选后的OTU表中的特征数量。
方法2:按数量/计数筛选。首先按丰度排序,默认为降序排序(从大到小):
usearch -otutab_sortotus ../otutab_rare.txt \
-output otutab_sort.txt
运行以下代码,提取指定数量的高丰度OTU,例如前100个:
sed '1 s/#OTU ID/OTUID/' otutab_sort.txt \
| head -n101 > otutab.txt
筛选完成后执行sed -i '1 s/#OTU ID/OTUID/' otutab.txt修改特征ID的列名。运行代码cut -f 1 otutab.txt > otutab_high.id提取用于序列检索的ID。然后,通过运行以下代码筛选高丰度细菌/指定差异细菌对应的OTU序列:
usearch -fastx_getseqs ../otus.fa -labels otutab_high.id -fastaout otus.fa
head -n 2 otus.fa
基于物种注释筛选OTU:
awk 'NR==FNR{a[$1]=$0} NR>FNR{print a[$1]}' ../taxonomy.txt \
otutab_high.id > otutab_high.tax
获得与OTU相对应的组平均值,用于生成样本热图(依赖于之前otu_mean.R计算过的按Group分组的均值):
awk 'NR==FNR{a[$1]=$0} NR>FNR{print a[$1]}' ../otutab_mean.txt otutab_high.id \
| sed 's/#OTU ID/OTUID/' > otutab_high.mean
将物种注释和丰度合并到注释文件中:
cut -f 2- otutab_high.mean > temp
paste otutab_high.tax temp > annotation.txt
head -n 3 annotation.txt
构建进化树
起始文件是result/tree目录下的otus.fa(序列),annotation.txt(物种和相对丰度)。执行命令muscle -in otus.fa -out otus_aligned.fas调用软件Muscle进行序列比对。
方法1:使用IQ-TREE快速构建ML进化树(耗时2m)
rm -rf iqtree
mkdir -p iqtree
iqtree -s otus_aligned.fas -bb 1000 -redo -alrt 1000 -nt AUTO -pre iqtree/otus
方法2:在Ubuntu上使用apt install fasttree安装FastTree,非常适合大型系统发育树(数百个OTU)的构建。通过执行代码fasttree -gtr -nt otus_aligned.fas > otus.nwk构建树。
进化树美化
本节概述了如何自定义系统发育树的外观,重点关注物种注释和丰度信息的外圈着色以及塑造。利用之前我们生成的annotation.txt文件包含与OTU对应的物种注释和丰度值,可以使用以下几个方案来进行美化:
方案1:外圈的颜色、形状和丰度注释文件
cd ${wd}/result/tree
Rscript ${db}/script/table2itol.R -a -c double -D plan1 -i OTUID -l Genus -t %s -w 0.5 annotation.txt
方案2:丰度柱状图注释文件
Rscript ${db}/script/table2itol.R -a -d -c none -D plan2 -b Phylum -i OTUID -l Genus -t %s -w 0.5 annotation.txt
方案3:热图注释文件
Rscript ${db}/script/table2itol.R -c keep -D plan3 -i OTUID -t %s otutab.txt
方案4:将整数转换为因子以生成注释文件
Rscript ${db}/script/table2itol.R -a -c factor -D plan4 -i OTUID -l Genus -t %s -w 0 annotation.txt
完成方案文件之后访问网站http://itol.embl.de/,在分析结果的“tree”文件夹中找到需要美化的进化树文件(通常命名为“otus.contree”)并将该文件拖放到iTOL上传区域。同时上传方案文件(plan1/2/3/4)进一步定制树形(例如,突出显示特定的分支)。iTOL可通过各种选项来调整树的外观,用户可根据喜好更改选项从而美化进化树。美化完成后,将进化树保存在iTOL中(图 4F)。或者用户可前往另一个在线可视化平台https://www.bic.ac.cn/BIC/#/,同样用户友好。
Linux下的附加分析(可选)
EasyAmplicon的这一部分提供了在Linux服务器上运行LEfSe、FAPROTAX和PICRUSt函数等分析的说明。熟悉命令行操作的用户会发现这些步骤很简单。但是,需要安装特定的软件,包括Linux系统、Conda包和QIIME2。虽然PICRUSt2是预测宏基因组功能内容的可选工具,但需要注意的是它资源较大。在开始之前,请确保您的终端从“Git Bash”切换到“在Linux下的Windows子系统(Windows Subsystem for Linux)”。请记住,在Linux上运行分析可能需要具有足够内存的服务器环境(>16GB)。为了可视化LEfSe分析,建议使用前面提到的在线网络服务器。
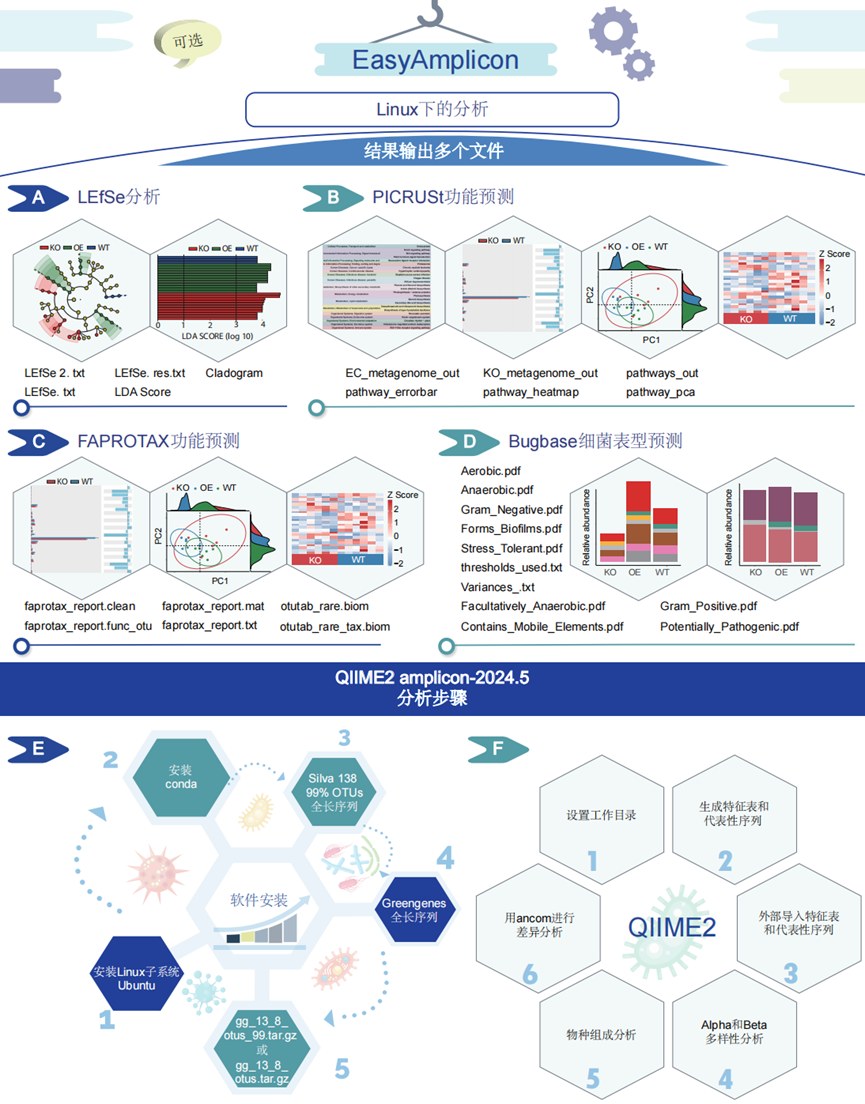
图5. EasyAmplicon在Linux系统下以及与QIIME 2集成的分析
(A)LEfSe是一种微生物组分析方法,用于识别群体间差异丰度的生物标志物。LEfSe分析利用LEfSe.txt、LEfSe.res.txt和otutab_rare.biom等输入文件。(B)PICRUSt功能预测根据标记基因测序数据预测微生物群落的功能组成,输出文件如EC_metagenome_out、KO_metagenome_out和pathways_out。(C)FAPROTAX功能预测根据标记基因数据推断微生物群落的功能概况,进行输出,生成包括faptotax_report.txt、faptotax_report.func_otu和faptotax_report.mat。(D)BugBase是一种细菌表型预测工具,它分析微生物组数据以预测细菌表型,生成各种PDF和文本文件,例如aerobic.pdf、anaerobic.pdf、forms_biofilms.pdf、gram_negative.pdf、gram_positive.pdf、facultatively_anaerobic.pdf、contains_mobile_elements.pdf、potentially_pathogenic.pdf、stress_tolerant.pdf、thresholds_used.txt和variances.txt。(E)QIIME 2分析设置从安装和激活Conda和各种数据库(包括Silva 138 99%和Greengenes)开始。(F)QIIME 2分析工作流程从设置工作目录开始。接下来,它会生成特征表,总结样本中生物特征的丰度,并选择代表性序列来代表每个OTU。可选的外部导入允许用户将从其他软件生成的数据导入QIIME 2进行进一步分析。然后,利用Alpha和Beta多样性分析来评估微生物群落内部和之间的多样性。之后,物种注释使用Greengenes等参考数据库将分类信息分配给OTU或者特征表中的ASV。最后,差异丰度分析使用ANCOM来识别样本组之间差异丰度的特征。
LEfSe分析
LEfSe识别样本中不同组之间具有统计学显著差异的特征。运行如下命令为LEfSe的输出创建目录:
mkdir -p ~/EasyAmplicon/lefse
cd ~/EAsyAmplicon/lefse
接下来,通过运行此代码在Linux系统下安装LEfSe(如果之前没有安装)conda install lefse。随后,运行lefse_format_input.py LEfSe.txt sample.in -u 1 -c 1 -o 1000000,将数据转换为LEfSe内部格式。调整1000000以指定要分析的最大特征数量。通过运行代码运行lefse_run.py sample.in input.res。接下来,通过执行以下代码绘制物种树来注释差异:
lefse_plot_cladogram.py input.res cladogram.pdf --format pdf
同样,要绘制所有差异特征的直方图,使用以下命令:
lefse_plot_res.py input.res res.pdf --format pdf
要绘制单个特征的柱状图(类似于STAMP中的条形图),请使用以下命令:
head input.res
lefse_plot_features.py -f one --feature_name "Bacteria.Firmicutes.Bacilli.Bacillales.Planococcaceae.Paenisporosarcina" \
--format pdf sample.in input.res Bacilli.pdf
此外,为了批量绘制所有差异特征柱状图,用户可以运行此代码:
mkdir -p features
lefse_plot_features.py -f diff --archive none --format pdf \
sample.in input.res features/
PICRUSt功能预测
PICRUSt可基于已测基因序列预测微生物群落的功能。对于可以访问Linux服务器的用户,以下步骤展示了PICRUSt的本地运行步骤。首先,设置数据库目录,以便从指定的URL进行下载。
db=/mnt/c/EasyMicrobiome
cd $db/gg
wget -c http://www.imeta.science/db/amplicon/GreenGenes/ko_13_5_precalculated.tab.gz
下一步涉及设置Conda环境。要创建并激活Conda环境,请执行以下代码:
n=picrust
conda create -n ${n} ${n} -c bioconda -y
wd=/mnt/c/EasyAmplicon
cd $wd/result/gg
conda activate picrust
下一步是数据准备。导航到包含Greengenes格式的处理后的OTU表(otutab.txt)的目录。通过运行以下代码,使用biom convert命令将OTU表转换为BIOM格式:
biom convert -i otutab.txt \
-o otutab.biom \
--table-type="OTU table" --to-json
要执行功能预测,请定义包含PICRUSt参考文件的数据库目录(例如/mnt/c/db)。要按拷贝数标准化OTU表,请运行以下代码:
db=/mnt/c/EasyMicrobiome
normalize_by_copy_number.py -i otutab.biom \
-o otutab_norm.biom \
-c ${db}/gg/16S_13_5_precalculated.tab.gz
为了预测宏基因组KEGG Orthology(KO)表,运行以下代码,该代码生成biom格式文件ko.biom(方便下游分类)和以制表符分隔的文本文件ko.txt(方便查看分析)。这里使用ko_13_5_precalculated.tab.gz数据库来预测宏基因组KO表。
predict_metagenomes.py -i otutab_norm.biom \
-o ko.biom \
-c ${db}/gg/ko_13_5_precalculated.tab.gz
predict_metagenomes.py -f -i otutab_norm.biom \
-o ko.txt \
-c ${db}/gg/ko_13_5_precalculated.tab.gz
要按级别对功能进行分类和汇总),处理ko.txt进行下游分析:
sed -i '/# Constru/d;s/#OTU //' ko.txt
num=`head -n1 ko.txt|wc -w`
paste <(cut -f $num ko.txt) <(cut -f 1-$[num-1] ko.txt) > ko.spf
for i in 1 2 3; do
categorize_by_function.py -f -i ko.biom -c KEGG_Pathways -l ${i} -o pathway${i}.txt
sed -i '/# Const/d;s/#OTU //' pathway${i}.txt
paste <(cut -f $num pathway${i}.txt) <(cut -f 1-$[num-1] pathway${i}.txt) > pathway${i}.spf
done
wc -l *.spf
FAPROTAX
本节概述了使用EasyAmplicon流程执行FAPROTAX分析所涉及的步骤。所需的输入文件包括txt格式的OTU表(可选,最好使用biom格式)和与OTU表关联的分类文件。输出将是:faprotax.txt(每个OTU的功能预测结果),faprotax_report.txt(关于OTU功能类别分配的详细报告),faprotax_report.mat(汇总功能注释的矩阵)。第一步是创建一个工作目录,脚本目录(sd)应指向FAPROTAX脚本在数据库目录中的存储位置。运行此代码:
wd=/mnt/c/EasyAmplicon/result/faprotax/
mkdir -p ${wd} && cd ${wd}
sd=/mnt/c/EasyMicrobiome/script/FAPROTAX_1.2.10
其次是软件安装,FAPROTAX在QIIME 2环境下运行可满足依赖关系,因此需要配置适当的QIIME 2环境(有关配置说明,请参阅QIIME 2部分)。要确定FAPRATOX功能并检查FAPROTAX是否安装正确,请运行:
python $sd/collapse_table.py
接下来,准备输入的OTU表,通过执行biom convert -i ../otutab_rare.txt -o otutab_rare.biom --table-type="OTU table" --to-json
将TXT格式的OTU表转换为biom json格式。然后,要将物种注释添加到biom表中,请运行以下代码:
biom add-metadata -i otutab_rare.biom --observation-metadata-fp ../taxonomy2.txt \
-o otutab_rare_tax.biom --sc-separated taxonomy --observation-header OTUID,taxonomy
要进行FAPROTAXS功能预测,请运行以下代码:
python ${sd}/collapse_table.py -i otutab_rare_tax.biom \
-g ${sd}/FAPROTAX.txt \
--collapse_by_metadata 'taxonomy' -v --force \
-o faprotax.txt -r faprotax_report.txt
最后,要生成功能注释矩阵,请运行以下代码:
grep 'ASV_' -B 1 faprotax_report.txt | grep -v -P '^--$' > faprotax_report.clean
根据此分析生成的输出结果列于图5A中。
注意:FAPROTAX已预先下载在EasyAmplicon脚本目录中。如果需要较新版本(例如此处使用的版本为1.2.10),请自主下载并将其放在脚本目录中。并确保将EasyMicrobiome/script/FAPROTAX_1.2.XX的路径替换为系统上的实际路径。
Bugbase细菌表型预测
本节详细介绍了在Linux环境下使用EasyAmplicon中的BugBase进行细菌表型预测的步骤。第一步是软件安装,BugBase已经预先整合到EasyMicrobiome中,需要更新原始代码才能运行。通常有两个方法,第一个是推荐的方法,第二个只需要运行一次,可自行选择。第一种方法需要Git下载(git clone https://github.com/knights-lab/BugBase)。第二种方法需要通过运行以下代码手动下载。
wget -c https://github.com/knights-lab/BugBase/archive/master.zip
mv master.zip BugBase.zip
unzip BugBase.zip
mv BugBase-master/ BugBase
cd BugBase
安装依赖项:
export BUGBASE_PATH=`pwd`
export PATH=$PATH:`pwd`/bin
run.bugbase.r –h
第二步,根据Greengene OTU表(本地分析支持txt格式,无需转换)和映射文件(在metadata.txt第一行添加#)准备“biom格式”输入文件。运行以下代码:
cd ~/EasyAmplicon/result
biom convert -i gg/otutab.txt -o otutab_gg.biom --table-type="OTU table" --to-json
sed '1 s/^/#/' metadata.txt > MappingFile.txt
使用Bugbase进行本地分析。
export BUGBASE_PATH=`pwd`
export PATH=$PATH:`pwd`/bin
run.bugbase.r -i otutab_gg.txt -m MappingFile.txt -c Group -o phenotype/
“-i”表示 OTU 表(otutab_gg.txt)的路径,“-m”表示映射文件(MappingFile.txt)的路径,“-c”表示映射文件中包含组信息的列名(Group),“-o”表示表型预测结果的输出目录(phenotype/)。分析结果显示在图5A 中,标题为Bugbase细菌表型预测。
注意:上述分析生成的输出文件和图表,包括LEFSe、Picrust、FAPROTAX和Bugbase,分别如图5A、B、C和D所示。
Silme2随机森林/Adaboost
本节详细介绍了在EasyAmplicon流程中使用Silme2执行机器学习分类的过程。Silme2支持用于微生物组分析的随机森林和AdaBoost算法。第一步是软件安装和依赖项。此处提供的说明仅供参考。建议查阅Silme2说明手册以了解最新的安装过程。
cd ~/software/ # Replace with your desired download directory
wget https://github.com/swo/slime2/archive/master.zip
mv master.zip slime2.zip
unzip slime2.zip
mv slime2-master/ slime2
cp slime2/slime2.py ~/bin/ # Replace with your desired bin directory
chmod +x ~/bin/slime2.py
sudo pip3 install --upgrade pip
sudo pip3 install pandas
sudo pip3 install sklearn
接下来,在QIIME 2环境下运行Silme2分析。通过运行此代码激活QIIME 2环境“conda activate qiime2-2024.5”(确保您使用的是正确的QIIME 2版本) 。导航到Silme2脚本目录:“cd /mnt/d/EasyMicrobiome/script/slime2”(替换为您的实际脚本路径)。为了进行标准化,请运行“./slime2.py otutab.txt design.txt --normalize --tag ab_e4 ab -n 10000”。“otutab.txt”以txt格式表示用户OTU表的路径。“design.txt”表示包含样本元数据和结果变量的设计文件的路径。“--normalize”表示规范化数据(推荐)。“--tag ab_e4”表示AdaBoost结果的输出标签(例如,对于经过10000次迭代的AdaBoost,输出标签为ab_e4)。“ab”表示分类感兴趣的特征(替换为您的实际特征名称)。“-n 10000”表示AdaBoost的迭代次数(建议使用数百万次以获得稳健的模型)。随后,要使用RandomForest并在短时间内计算10,000次(为了稳健性,建议使用数百万次迭代,支持多线程),请运行此代码:
./slime2.py otutab.txt design.txt --normalize --tag rf_e4 rf -n 10000
PICRUSt2环境导入与导出
PICRUSt2扩展了原始PICRUSt的功能,以基于标记基因测序谱预测微生物群落的功能。第一步是安装PICRUSt2(https://github.com/picrust/picrust2),有几种安装方法可供选择。
方法1:
n=picrust2
conda create -n ${n} -c bioconda -c conda-forge ${n}=2.3.0_b
conda activate ${n}
方法2:导出安装环境cd ~/db/conda/,设置环境名称n=picrust2。然后,运行以下代码来激活picrust同时打包压缩包。
conda activate ${n}.
conda pack -n ${n} -o ${n}.tar.gz
方法3:导入安装环境。如Qiime2和humann2(含picurst),通过运行代码“n=picrust2”设置环境名称。接下来,使用以下命令下载环境包:
wget -c ftp://download.nmdc.cn/tools/conda/${n}.tar.gz
指定安装目录并解压包:
condapath=~/miniconda3
mkdir -p ${condapath}/envs/${n}
tar -xvzf ${n}.tar.gz -C ${condapath}/envs/${n}
最后,通过运行此代码激活并初始化环境:
source ${condapath}/envs/${n}/bin/activate
conda unpack
PICRUSt2功能预测
激活PICRUSt2环境后,从Conda激活开始,逐步运行以下代码。要加载picrust2环境,请运行代码“conda activate picrust2”。然后,运行以下命令创建名为“picrsut2”的目录。
wd=/mnt/c/EasyAmplicon/result/picrust2
mkdir -p ${wd} && cd ${wd}
然后执行此命令:
picrust2_pipeline.py -s ../otus.fa -i ../otutab.txt -o ./out -p 8
此过程利用OTU序列文件“../otus.fa”、OTU表和“../otutab.txt”。大约需要12分钟才能完成。然后,通过执行以下代码添加EC/KO/Pathway注释:
cd out
add_descriptions.py -i pathways_out/path_abun_unstrat.tsv.gz -m METACYC \
-o pathways_out/path_abun_unstrat_descrip.tsv.gz
add_descriptions.py -i EC_metagenome_out/pred_metagenome_unstrat.tsv.gz -m EC \
-o EC_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gz
add_descriptions.py -i KO_metagenome_out/pred_metagenome_unstrat.tsv.gz -m KO \
-o KO_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gz
要按层次结构合并KEGG,请运行给定的代码:
db=/mnt/c/EasyMicrobiome/
zcat KO_metagenome_out/pred_metagenome_unstrat.tsv.gz > KEGG.KO.txt
python3 ${db}/script/summarizeAbundance.py \
-i KEGG.KO.txt \
-m ${db}/kegg/KO1-4.txt \
-c 2,3,4 -s ',+,+,' -n raw \
-o KEGG
最后,为了计算每个级别的特征数量,运行代码“wc -l KEGG*”:
注意:此分析需要大量资源,可能需要具有足够内存的服务器环境(>16GB)。
QIIME 2流程下的分析
EasyAmplicon与QIIME 2无缝集成,后者是一种用于分析微生物组测序数据的流行生物信息学平台。本节提供了在EasyAmplicon工作流程中使用QIIME 2的基本指南。QIIME 2主要针对Linux和Mac系统设计。Windows用户可以利用WSL或Linux服务器环境。
视频教程链接:https://youtu.be/PghGX9zXqm0
QIIME 2安装
EasyAmplicon提供了清晰的说明和可预配置的环境来进行QIIME 2安装(图 5E)。要继续使用Qiime2,请导航到EasyAmplicon master/qiime2目录,使用R studio打开“qiime2_pipeline”。接下来,使用WSL打开一个新终端,并通过运行以下代码激活现有的Conda环境:
n=qiime2-amplicon-2024.5
wget -c https://data.qiime2.org/distro/amplicon/${n}-py39-linux-conda.yml
conda env create -n ${n} --file ${n}-py39-linux-conda.yml
conda pack -n ${n} -o ${n}.tar.gz
conda activate ${n}
注意:有关Conda的安装,请参阅“下载和安装 Conda ”部分以获取详细说明。
下载物种注释数据
我们需要下载两个预先训练的参考数据库用于物种注释(图 5E),包括Silva 138 99% OTU全长序列和Greengenes2 2022.10或更高版本的全长序列。用户可以从QIIME 2官方网站下载所需的数据库。要下载Silva 138,请运行此代码:
wget -c https://data.qiime2.org/202 4 .2/common/silva-138-99-nb-classifier.qza
对于Greengenes2,运行以下代码:
wget -c https://data.qiime2.org/2024.2/common/silva-138-99-nb-classifier.qza
训练分类器
本节以Greengenes为例,演示如何使用下载的参考数据库训练自定义分类器。首先,通过运行以下代码来设置工作目录:
wd=/mnt/c/EasyAmplicon/qiime2
mkdir -p $wd
cd $wd
要下载Greengenes参考数据库(还提供了一个较小的99% OTU数据库),请运行以下代码:
wget -c ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
wget -c ftp://download.nmdc.cn/tools/amplicon/GreenGenes/gg_13_8_otus_99.tar.gz
mv gg_13_8_otus_99.tar.gz gg_13_8_otus.tar.gz
解压下载的存档文件:
tar -zxvf gg_13_8_otus.tar.gz
通过运行下面的命令导入参考序列,将引用序列从存档文件(gg_13_8_otus)的“rep_set”目录下的“99_otus.fasta”文件导入到名为“99_otus.qza”的QIIME工件中。
qiime tools import --type 'FeatureData[Sequence]' --input-path gg_13_8_otus/rep_set/99_otus.fasta --output-path 99_otus.qza
然后,要将物种注释信息从gg_13_8_otus分类目录下的99_OTU_taxonomy.txt文件导入到名为ref-taxonomy.qza的QIME工件中:
qiime tools import --type 'FeatureData[Taxonomy]' --input-format HeaderlessTSVTaxonomyFormat --input-path gg_13_8_otus/taxonomy/99_otu_taxonomy.txt --output-path ref-taxonomy.qza
为了训练名为classifier_gg_13_8_99.qza的全长分类器(可能需要30分钟),通过“--i-reference-reads”指定导入的引用序列(99_otus.qza),“--i-reference-taxonomy”指定导入的注释信息:
time qiime feature-classifier fit-classifier-naive-bayes --i-reference-reads 99_otus.qza --i-reference-taxonomy ref-taxonomy.qza --o-classifier classifier_gg_13_8_99.qza
根据所提供的正向(AACMGGATTAGATACCCKG)和反向(ACGTCATCCCCACCTTCC)引物提取扩增片段,这可能需要8分钟。提取的序列存储在名为ref-seqs.qza的QIIME工件中。记得用实验中实际使用的引物代替这些引物。
time qiime feature-classifier extract-reads --i-sequences 99_otus.qza --p-f-primer AACMGGATTAGATACCCKG --p-r-primer ACGTCATCCCCACCTTCC --o-reads ref-seqs.qza
要使用提取的片段训练分类器(可能需要7分钟),请运行以下代码:
time qiime feature-classifier fit-classifier-naive-bayes --i-reference-reads ref-seqs.qza --i-reference-taxonomy ref-taxonomy.qza --o-classifier classifier_gg_13_8_99_V5-V7.qza
Qiime2基本工作流程
一旦所有软件成功安装,用户就可以开始使用QIIME 2流程进行基本分析(图 5F)。
准备工作
首先通过运行以下代码来设置工作目录:
wd=/mnt/c/Easyamplicon/qiime2/
mkdir -p ${wd}
cd ${wd}
激活QIIME2环境(如果您使用不同的版本,请将Qiime2版本替换为您的特定版本)。
conda activate qiime2-amplicon-2024.5
准备样本元数据文件(metadata.txt)和原始序列数据文件(seq/*.fq.gz)。
ln /mnt/c/EasyAmplicon/seq/* seq/
ln /mnt/c/EasyAmplicon/result/metadata.txt ./
根据您的元数据生成一份清单文件,列出样本 ID、正向和反向FASTQ文件路径。
awk 'NR==1{print "sample-id\tforward-absolute-filepath\treverse- absolute-filepath"} NR>1{print $1"\t"$PWD/seq/"$1"_1.fq.gz\t"$PWD/seq/"$1"_2.fq.gz"}' metadata.txt > manifest
head -n3 manifest
将您的数据导入QIIME2,格式为双端33。
qiime tools import --type 'SampleData[PairedEndSequencesWithQuality]' --input-path manifest --output-path demux.qza --input-format PairedEndFastqManifestPhred33V2
生成特征表和代表序列
QIIME2中生成特征表和代表序列主要有两种方式:
方法1:DADA2(可选)。此方法使用DADA2执行去噪,但与推荐方法相比,它可能更慢且更容易出错。
time qiime dada2 denoise-paired --i-demultiplexed-seqs demux.qza --p-n-threads 4 --p-trim-left-f 29 --p-trim-left-r 18 --p-trunc-len-f 0 --p-trunc-len-r 0 --o-table dada2-table.qza --o-representative-sequences dada2-rep-seqs.qza --o-denoising-stats denoising-stats.qza
方法2:外部导入。这是推荐的方法,允许用户从其他软件导入预先生成的特征表(例如OTU表)和代表性序列。为了将OTU表(otutab.txt)转换为biom格式,请运行以下命令:
biom convert -i otutab.txt -o otutab.biom --table-type="OTU table" --to-json
要将特征表导入QIIME2 ,请运行以下代码:
qiime tools import --input-path otutab.bi--type 'FeatureTable[Frequency]' --input-format BIOMV100Format --output-path table.qza
要导入代表序列,请运行下面给出的代码:
qiime tools import --input-path otus.fa --type 'FeatureData[Sequence]' --output-path rep-seqs.qza
导入后,使用这些命令生成汇总统计数据:
qiime feature-table summarize --i-table table.qza --o-visualization table.qzv --m-sample-metadata-file metadata.txt
qiime feature-table tabulate-seqs --i-data rep-seqs.qza --o-visualization rep-seqs.qzv
Alpha和Beta多样性分析
这一步将会构建一个系统发育树,用于展示数据集中微生物序列之间进化关系。它输入代表性序列(rep-seqs.qza),并输出对齐序列(aligned-rep-seqs.qza)、屏蔽歧义对齐区域的屏蔽序列(masked-aligned-rep-seqs.qza)、无根系统发育树(unrooted-tree.qza)、有根系统发育树(rooted-tree.qza)。运行命令:
qiime phylogeny align-to-tree-mafft-fasttree --i-sequences rep-seqs.qza --o-alignment aligned-rep-seqs.qza --o-masked-alignment masked-aligned-rep-seqs.qza --o-tree unrooted-tree.qza --o-rooted-tree rooted-tree.qza
通过运行以下代码计算特定采样深度下的核心多样性指标,采样深度通常选择最小值:
qiime diversity core-metrics-phylogenetic --i-phylogeny rooted-tree.qza --i-table table.qza --p-sampling-depth 7439 --m-metadata-file metadata.txt --output-dir core-metrics-results
分析和可视化元数据文件中定义的组之间的Alpha多样性差异。用户可以选择不同的Alpha多样性指数(以observed_features为例):
index=observed_features
qiime diversity alpha-group-significance --i-alpha-diversity core-metrics-results/${index}_vector.qza --m-metadata-file metadata.txt --o-visualization core-metrics-results/${index}-group-significance.qzv
为了生成Alpha样性稀释曲线来直观地展示多样性如何随测序深度而变化,请运行以下代码:
qiime diversity alpha-rarefaction --i-table table.qza --i-phylogeny rooted-tree.qza --p-max-depth 10298 --m-metadata-file metadata.txt --o-visualization alpha-rarefaction.qzv
使用选定的指数(例如,weighted_unifrac)和PERMANOVA检验,分析并可视化组之间的Beta多样性差异。
distance=weighted_unifrac
column=Group
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/${distance}_distance_matrix.qza \
--m-metadata-file metadata.txt \
--m-metadata-column ${column} \
--o-visualization core-metrics-results/${distance}-${column}-significance.qzv \
--p-pairwise
物种组成分析
本节指导用户使用预先训练的分类器(例如 SILVA 数据库)对序列进行分类。
time qiime feature-classifier classify-sklearn \
--i-classifier classifier_gg_13_8_99.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
为了使用QIIME2可视化来直观显示样本的分类组成,请运行以下代码:
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
使用堆叠柱状图显示数据:
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file metadata.txt \
--o-visualization taxa-bar-plots.qzv
基于ANCOM进行差异特征分析
为了将伪计数添加到特征表中以进行稳健的统计分析,请运行以下代码:
qiime composition add-pseudocount \
--i-table table.qza \
--o-composition-table comp-table.qza
要使用ANCOM识别组之间的差异丰度特征,请运行以下命令:
column=Group
time qiime composition ancom \
--i-table comp-table.qza \
--m-metadata-file metadata.txt \
--m-metadata-column ${column} \
--o-visualization ancom-${column}.qzv
也可以通过合并特征表和重复ANCOM分析,在更高的分类水平(例如,属)上进行差异分析:
qiime taxa collapse \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--p-level 6 \
--o-collapsed-table table-l6.qza
通过运行此代码来格式化特征表并添加伪计数:
qiime composition add-pseudocount \
--i-table table-l6.qza \
--o-composition-table comp-table-l6.qza
为了计算差异属,运行以下代码并指定用于比较的分组类型:
qiime composition ancom \
--i-table comp-table-l6.qza \
--m-metadata-file metadata.txt \
--m-metadata-column ${column} \
--o-visualization ancom-l6-${column}.qzv
预期结果
EasyAmplicon流程提供了每个步骤的脚本和说明,并可引用外部资源,易于使用和适应。它支持多样性指数的批量处理,自动计算并生成可视化图标,如箱线图、柱状图、稀释曲线和Alpha多样性的维恩图等。还可使用距离矩阵和PCoA图等分析Beta多样性。它使用参考数据库生成堆叠柱状图、弦/圈图和树状图来展示物种的功能预测和组成。LEfSe识别差异丰度特征,而PICRUSt2预测功能途径。FAPROTAX分析OTU在元素循环中的作用。使用EasyAmplicon流程为全面分析微生物组样本(植物、动物、土壤、环境)的组成和多样性提供了宝贵的见解,为进一步探索任何宿主-微生物相互作用铺平了道路。这个详细的实验流程旨在使各个领域的研究人员能够更轻松地深入研究微生物组,最终扩大微生物组研究的范围和影响。虽然EasyAmplicon主要针对第二代测序数据设计,但未来的更新旨在引入新功能从而提高与第三代测序平台的兼容性。
结论
本文介绍的EasyAmplicon方案为研究人员提供了全面而详细的指南,帮助他们高效、有效地进行扩增子分析。通过将多种流行的生物信息学工具集成到简化的工作流程中,大大降低了通常与高通量数据分析相关的技术障碍。该方案详细解释了从数据预处理和多样性分析到高级可视化技术的每个步骤,确保用户无需具备丰富的生物信息学专业知识即可进行操作。
代码和数据可用性
EasyAmplicon v1.0数据分析流程的计算脚本可在GitHub上免费获取,网址为https://github.com/YongxinLiu/EasyAmplicon。EasyAmplicon网络服务器也可免费获取,并定期更新最新版本的软件和数据库。我们鼓励用户参考GitHub以获取与EasyAmplicon流程相关的更新链接。补充材料可通过DOI或iMeta Science http://www.imeta.science/找到。
引文格式:
ousuf, Salsabeel, Hao Luo, Meiyin Zeng, Lei Chen, Tengfei Ma, Xiaofang Li, Maosheng Zheng, et al. “Unveiling microbial communities with EasyAmplicon: A user-centric guide to perform amplicon sequencing data analysis.” iMetaOmics: e42. https://doi.org/10.1002/imo2.42
附图

作者简介

Salsabeel Yousuf(第一作者)
● 中国农业科学院深圳农业基因组研究所博士后。
● 研究横跨动物科学、育种、繁殖、遗传学和微生物学。在iMetaOmics、Cells、Ovarian research等著名SCI期刊以第一作者(含合著者)发表SCI论文15篇。

罗豪(第一作者)
● 中国农业科学院深圳农业基因组研究所,在读博士研究生。
● 研究方向为人工智能在微生物组数据分析中的应用,在iMeta、iMetaOmics、GigaScience、Journal of Hazardous Materials等期刊发表论文5篇,其中第一/共同第一作者3篇。

曾美尹(第一作者)
● 中国农业科学院深圳农业基因组研究所在读硕士研究生,毕业于华北理工大学生命科学学院。

刘永鑫(通讯作者)
● 中国农业科学院深圳农业基因组研究所研究员,博士生导师。2014年博士毕业于中国科学院大学生物信息学专业,之后在中国科学院遗传与发育生物学研究所工作历任博士后、工程师、高级工程师,2022年10月加入中国农业科学院深圳农业基因组研究所担任课题组长。
● 研究方向为宏基因组方法开发、功能挖掘和科学传播。参与QIIME 2项目,主导开发了易扩增子(EasyAmplicon)、易宏基因组(EasyMetagenome)、培养组(Culturome)分析流程、数据分析网站(EVenn、ImageGP)和R包(amplicon、ggClusterNet)等,目标是全面打造宏基因组领域方法学基础设施,推动微生物组学发展。以(共同)第一或通讯作者在Nature Biotechnology、Nature Microbiology、iMeta等期刊发表论文20余篇。合作在Science、Cell Host & Microbe、Microbiome等期刊发表论文20余篇,累计发表论文50余篇,被引用13000+次。主编《微生物组实验手册》专著,由300多位同行参与,共同打造本领域长期更新的中文百科全书。创办宏基因组公众号,15万+同行关注,分享原创文章3千余篇,累计阅读量超4千万,打造本领域最具影响力的科学传播平台。发起iMeta期刊,联合全球千位专家共同打造宏基因组学、微生物组和生物信息学顶刊,解决我国本领域期刊出版卡脖子问题。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作
● iMetaOmics | 中科院动物所金坚石组综述16S rRNA基因扩增子测序技术的“前世今生”
● iMetaOmics | 浙大张天真组完成二倍体棉种泛基因组构建
● iMetaOmics | 张勇/李福平-先进糖蛋白组学在男性生殖研究中的潜在应用
● iMetaOmics | 暨南大学潘永勤/杨华组-炎症蛋白联合检测利于诊断甲状腺乳头状癌和结节性甲状腺肿
● iMetaOmics | 张开春组利用多组学方法揭示甜樱桃加倍后果色变化的候选基因
● iMetaOmics | 杜娟/林婷婷-慢性泪囊炎患者眼部菌群类型和纵向菌群变化
● iMetaOmics | 陈汉清/陈俊综述有关肝细胞癌治疗的新兴纳米医学策略
● iMetaOmics | 基因组所刘永鑫/卢洪评述微生物在提高杂种优势中的作用
● iMetaOmics | 上科大刘雪松组开发基于通路的肿瘤细胞鉴别工具TCfinder
● iMetaOmics | 中山大学刘鹏/邹宇田-整合人工智能实现HER2阳性乳腺癌精准管理
● iMetaOmics | 安徽农大李晓玉组-丛枝菌根真菌对玉米内生菌群的影响
● iMetaOmics | 徐涛/黄蓉/苏国海-急性冠脉综合征纵向多组学队列建设
● iMetaOmics | 通过整合宏组学促进人类与环境健康发展
● iMetaOmics | 苏州大学林俊组-揭示活性微生物及益生元/益生菌与关节炎联系
● iMetaOmics | 中国药科大学徐文波开发叶绿体基因组数据分析软件
● iMetaOmics | 清华刘晓组和复旦王久存组揭示特定细菌在皮肤老化中的作用
●iMetaOmics | 中南大学夏晓波团队揭示青光眼和SLE发病机制新关联
●iMetaOmics | 庐山植物园刘芬组揭示了自噬在植物-根微生物互作机制中的调控作用
●iMetaOmics | 杨瑞馥/袁静综述微生物组与“同一健康”的联系
●iMetaOmics | 同济/上海交大-开发支持群体分组分析的宏基因组测序综合分析软件
●iMetaOmics | 陈绍鸣-关于靶向NF-κB的潜伏逆转剂及其在HIV潜伏期的表观遗传和突变影响的评论
● iMetaOmics | 甘肃农大刘自刚组-强抗寒甘蓝型冬油菜的基因组组装和基因组特征解析
● iMetaOmics | 南京农大朱伟云组-外周血清素在结肠稳态中的作用
● iMetaOmics | 魏来/贾慧珏/何明光-多组学助力揭示塑造转录组的基因型-微生物组相互作用
● iMetaOmics | 徐州医科大学朱作斌组-微生物对寿命的调节:机制和治疗策略
● iMetaOmics | 白立景/邢凯组-解析脊椎动物肠道微生物多样性的影响因素
● iMetaOmics | 刘永鑫/陈同-用于食物微生物组成和时间序列研究的微生物组数据库FoodMicroDB
● iMetaOmics | 重庆大学王贵学组-肠道微生物细胞外囊泡在神经退行性疾病中的新作用及其治疗策略
● iMetaOmics | 四川大学王红宁组-解析产气荚膜梭菌的基因组宿主适应性
● iMetaOmics | 北京协和医院杨启文组-ramR基因突变增强免疫激活和依拉环素耐药性
● iMetaOmics | 香港中文苏奇组-抗菌多肽开发中的见解: 一个多学科视角的观察
● iMetaOmics | 上科大刘雪松组开发CD4 TCR特异性预测工具Pep2TCR
● iMetaOmics | 江苏省农科院植物细菌团队-解析中国梨火疫菌特征及溯源分析
更多推荐
(▼ 点击跳转)
iMeta | 引用15000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐

 0
0 0
0- 0



 已为社区贡献55条内容
已为社区贡献55条内容

所有评论(0)