杂化泛函静态自洽scf/非自洽计算
杂化泛函计算步骤态密度、能带结构建议:1. 结构优化(PBE)2. 静态自洽scf计算(PBE)*保留波函数和电荷密度3. 静态自洽scf计算(HSE06)*采用第2步得到的波函数,加快收敛速度*保留波函数和电荷密度4. 非自洽计算(HSE06)*采用第3步得到的波函数和电荷密度
杂化泛函概述
Hartree-Fock理论包含精确的交换作用能(从KS轨道得到的HF交换被特别称为精确交换)。因此,在DFT中混合部分精确交换可能有助于提高精度(Jacobi之梯的第四阶)。但是,必须注意到直接将精确交换作为交换泛函的效果并不好,因为局域的关联部分需要一个误差抵消。最简单的杂化泛函。
可以写成:
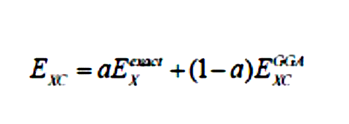
常用杂化泛函
B3LYP适用于分子体系
The standard hybrid functional for molecules after Becke, Lee, Yang& Parr47, Vosko, Wilk & Nusair, Stephens, Devlin, Chabalowski & Frisch
PBE0
25%的HF ,75%的DFT,适用于固体体系
The standard hybrid functional for solids developed by Ernzerhof &Scuseria and Adamo & Barone
HSE06
The hybrid functional developed by Heyd, Scuseria & Ernzerhof. PBE0基础上考虑了屏蔽库伦效应,适用于固体体系, 尤其带隙计算HSE06具有很好的收敛性。目前使用最为广泛。
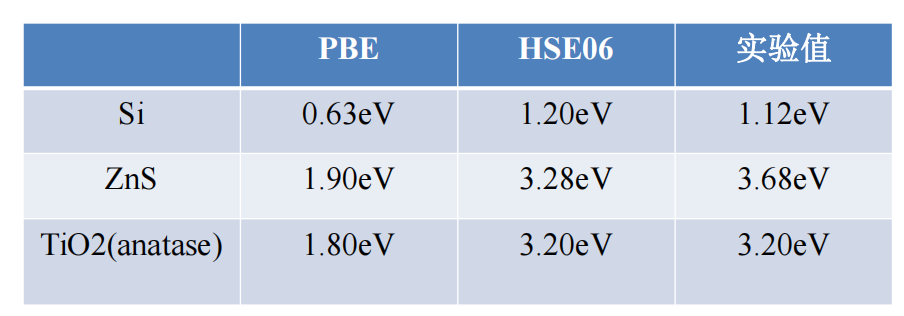
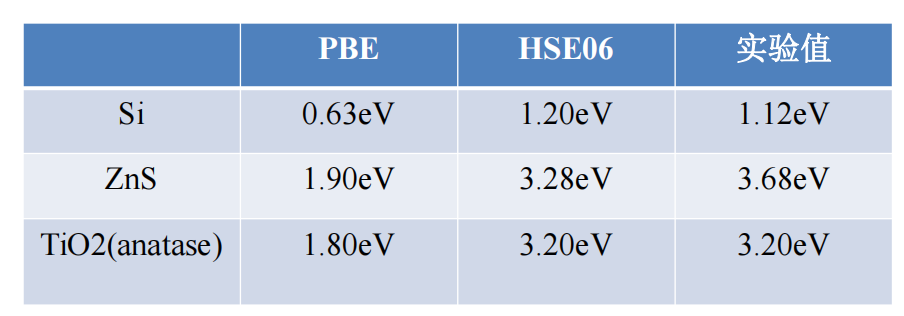
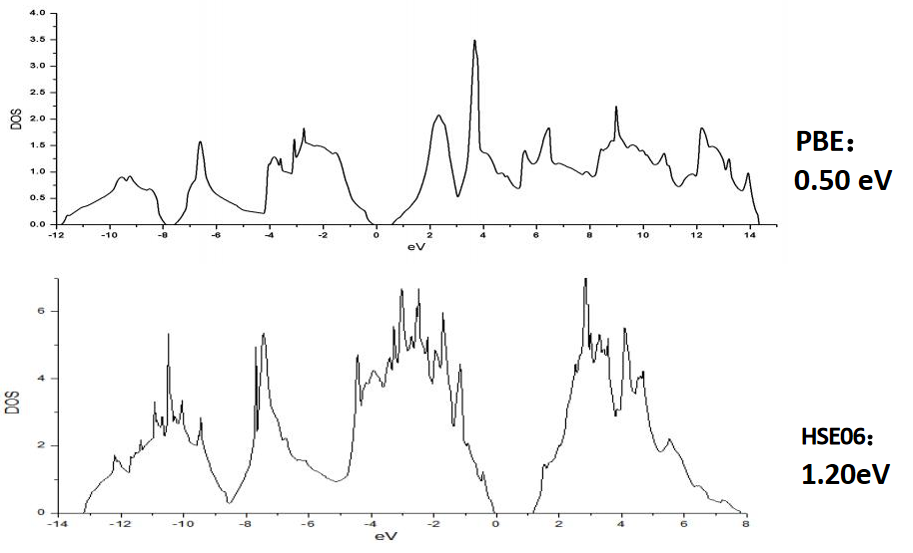
半导体材料GGA-PBE与HSE06泛函计算对比
半导体
禁带宽度是半导体十分重要的电子性质。
• 众所周知,普通的LDA和GGA方法计算带隙存在问题,一般低估1-2个电子伏特。解决的办法则是采用杂化泛函HSE给出更为准确的禁带宽度。
杂化泛函相关参数
LHFCALC = .TRUE.
*采用杂化泛函计算
HFSCREEN = 0.2
*0.2代表HSE06 ,0.3代表HSE03
PREC = Accurate
*用杂化计算,精度要高
LMAXFOCK = 4
*s/p轨道或过渡金属选4;f轨道需要选6
ALGO = Damped
*杂化计算时,采用Normal自洽很难收敛,Damped更容易收敛
TIME = 0.4
*杂化计算时,利于收敛。若未收敛,可适度降低。
PRECFOCK = Fast/Normal/Accurate
*需要计算速度快一点时,选择Fast,如果要求能量和力非常精确 ,可选择Normal和Accurate。
杂化泛函计算步骤
态密度、能带结构建议:
1. 结构优化(PBE)
2. 静态自洽scf计算(PBE)
*保留波函数和电荷密度
3. 静态自洽scf计算(HSE06)
*采用第2步得到的波函数,加快收敛速度
*保留波函数和电荷密度
4. 非自洽计算(HSE06)
*采用第3步得到的波函数和电荷密度
举例——Si
应用领域:
太阳能电池 -电子元器件-半导体等
禁带宽度:实验值为1.20eV
静态自洽scf (HSE06)
创建HSE-scf文件夹:
cp -r Si-scf Si-HSE-scf
修改输入文件:
INCAR (如右修改)
KPOINTS (不变)
POTCAR (不变)
POSCAR (不变)

非自洽计算(HSE06)
创建HSE-scf文件夹:
cp -r Si-HSE-scf Si-HSE-dos
修改输入文件:
INCAR (如右修改)
KPOINTS (不变)
POTCAR (不变)
POSCAR (不变)

结果分析

DAMO开发者矩阵,由阿里巴巴达摩院和中国互联网协会联合发起,致力于探讨最前沿的技术趋势与应用成果,搭建高质量的交流与分享平台,推动技术创新与产业应用链接,围绕“人工智能与新型计算”构建开放共享的开发者生态。
更多推荐

 3
3 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)